after_diagnosis_qc_gigamuga_12_batches
Hao He
2020-11-07
Last updated: 2020-11-07
Checks: 7 0
Knit directory: csna_workflow/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190918) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 553d37f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: analysis/.Rhistory
Untracked files:
Untracked: analysis/.nfs0000000135a08b9b00002448
Untracked: analysis/.nfs0000000135a08ba50000244a
Untracked: analysis/07_do_diversity_report.Rmd
Untracked: analysis/19_qtl_analysis_RTG20_GS_a1.R
Untracked: analysis/19_qtl_analysis_RTG20_GS_a1.err
Untracked: analysis/19_qtl_analysis_RTG20_GS_a1.out
Untracked: analysis/19_qtl_analysis_RTG20_GS_a1.sh
Untracked: analysis/19_qtl_analysis_RTG20_GS_a2.R
Untracked: analysis/19_qtl_analysis_RTG20_GS_a2.err
Untracked: analysis/19_qtl_analysis_RTG20_GS_a2.out
Untracked: analysis/19_qtl_analysis_RTG20_GS_a2.sh
Untracked: analysis/19_qtl_analysis_RTG20_GS_a3.R
Untracked: analysis/19_qtl_analysis_RTG20_GS_a3.err
Untracked: analysis/19_qtl_analysis_RTG20_GS_a3.out
Untracked: analysis/19_qtl_analysis_RTG20_GS_a3.sh
Untracked: analysis/19_qtl_analysis_RTG20_GS_a4.R
Untracked: analysis/19_qtl_analysis_RTG20_GS_a4.err
Untracked: analysis/19_qtl_analysis_RTG20_GS_a4.out
Untracked: analysis/19_qtl_analysis_RTG20_GS_a4.sh
Untracked: analysis/19_qtl_analysis_RTG20_G_a1.R
Untracked: analysis/19_qtl_analysis_RTG20_G_a1.err
Untracked: analysis/19_qtl_analysis_RTG20_G_a1.out
Untracked: analysis/19_qtl_analysis_RTG20_G_a1.sh
Untracked: analysis/19_qtl_analysis_RTG20_G_a2.R
Untracked: analysis/19_qtl_analysis_RTG20_G_a2.err
Untracked: analysis/19_qtl_analysis_RTG20_G_a2.out
Untracked: analysis/19_qtl_analysis_RTG20_G_a2.sh
Untracked: analysis/19_qtl_analysis_RTG20_G_a3.R
Untracked: analysis/19_qtl_analysis_RTG20_G_a3.err
Untracked: analysis/19_qtl_analysis_RTG20_G_a3.out
Untracked: analysis/19_qtl_analysis_RTG20_G_a3.sh
Untracked: analysis/19_qtl_analysis_RTG20_G_a4.R
Untracked: analysis/19_qtl_analysis_RTG20_G_a4.err
Untracked: analysis/19_qtl_analysis_RTG20_G_a4.out
Untracked: analysis/19_qtl_analysis_RTG20_G_a4.sh
Untracked: analysis/csna_image.sif
Untracked: analysis/scripts/
Untracked: analysis/temp/
Untracked: analysis/workflow_proc.R
Untracked: analysis/workflow_proc.sh
Untracked: analysis/workflow_proc.stderr
Untracked: analysis/workflow_proc.stdout
Untracked: code/reconst_utils.R
Untracked: data/69k_grid_pgmap.RData
Untracked: data/FinalReport/
Untracked: data/GCTA/
Untracked: data/GM/
Untracked: data/Jackson_Lab_11_batches/
Untracked: data/Jackson_Lab_12_batches/
Untracked: data/Jackson_Lab_Bubier_MURGIGV01/
Untracked: data/Jackson_Lab_Gagnon/
Untracked: data/RTG/
Untracked: data/cc_variants.sqlite
Untracked: data/marker_grid_0.02cM_plus.txt
Untracked: data/mouse_genes_mgi.sqlite
Untracked: data/pheno/
Untracked: output/AfterQC_Percent_missing_genotype_data.pdf
Untracked: output/AfterQC_Proportion_matching_genotypes_after_removal_of_bad_samples.pdf
Untracked: output/AfterQC_Proportion_matching_genotypes_before_removal_of_bad_samples.pdf
Untracked: output/AfterQC_number_crossover.pdf
Untracked: output/DO_Gigamuga_chr2_WSB_G21.png
Untracked: output/DO_Gigamuga_chr2_WSB_G22.png
Untracked: output/DO_Gigamuga_chr2_WSB_G23.png
Untracked: output/DO_Gigamuga_chr2_WSB_G25.png
Untracked: output/DO_Gigamuga_chr2_WSB_G29.png
Untracked: output/DO_Gigamuga_chr2_WSB_G30.png
Untracked: output/DO_Gigamuga_chr2_WSB_G31.png
Untracked: output/DO_Gigamuga_chr2_WSB_G32.png
Untracked: output/DO_Gigamuga_chr2_WSB_G33.png
Untracked: output/DO_Gigamuga_founder_proportions_G21.png
Untracked: output/DO_Gigamuga_founder_proportions_G22.png
Untracked: output/DO_Gigamuga_founder_proportions_G23.png
Untracked: output/DO_Gigamuga_founder_proportions_G25.png
Untracked: output/DO_Gigamuga_founder_proportions_G29.png
Untracked: output/DO_Gigamuga_founder_proportions_G30.png
Untracked: output/DO_Gigamuga_founder_proportions_G31.png
Untracked: output/DO_Gigamuga_founder_proportions_G32.png
Untracked: output/DO_Gigamuga_founder_proportions_G33.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G21.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G22.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G23.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G25.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G29.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G30.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G31.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G32.png
Untracked: output/DO_Gigamuga_founder_proportions_chr2_G33.png
Untracked: output/DO_recom_block_size_G21.png
Untracked: output/DO_recom_block_size_G22.png
Untracked: output/DO_recom_block_size_G23.png
Untracked: output/DO_recom_block_size_G25.png
Untracked: output/DO_recom_block_size_G29.png
Untracked: output/DO_recom_block_size_G30.png
Untracked: output/DO_recom_block_size_G31.png
Untracked: output/DO_recom_block_size_G32.png
Untracked: output/DO_recom_block_size_G33.png
Untracked: output/Histgram_Prj01_RL-Acquisition_preqc_02142020.pdf
Untracked: output/Histgram_Prj01_RL-Reversal_preqc_02142020.pdf
Untracked: output/Histgram_Prj02_Sensitization_preqc_02142020.pdf
Untracked: output/Histgram_Prj04_IVSA_preqc_02142020.pdf
Untracked: output/Novelty_residuals_RankNormal_datarelease_12182019-69k-genelist.csv
Untracked: output/Novelty_residuals_RankNormal_datarelease_12182019-69k-variantlist.csv
Untracked: output/Novelty_residuals_RankNormal_datarelease_12182019-genelist.csv
Untracked: output/Novelty_residuals_RankNormal_datarelease_12182019-variantlist.csv
Untracked: output/Percent_genotype_errors_obs_vs_predicted.pdf
Untracked: output/Percent_missing_genotype_data.pdf
Untracked: output/Percent_missing_genotype_data_per_marker.pdf
Untracked: output/Proportion_matching_genotypes_after_removal_of_bad_samples.pdf
Untracked: output/Proportion_matching_genotypes_after_removal_samples_percent_missing_5.pdf
Untracked: output/Proportion_matching_genotypes_before_removal_of_bad_samples.pdf
Untracked: output/Proportion_matching_genotypes_before_removal_samples.pdf
Untracked: output/RTG/
Untracked: output/blup/
Untracked: output/condi.m2.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/condi.m2.qtl.blup_Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/condi.m2.qtl.out.RData
Untracked: output/conditional.69k.Novelty_residuals_RankNormal_datarelease_12182019-genelist.csv
Untracked: output/conditional.69k.Novelty_residuals_RankNormal_datarelease_12182019-variantlist.csv
Untracked: output/conditional.Novelty_residuals_RankNormal_datarelease_12182019-genelist.csv
Untracked: output/conditional.Novelty_residuals_RankNormal_datarelease_12182019-variantlist.csv
Untracked: output/conditional.m2.69k.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/conditional.m2.69k.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/conditional.m2.69k.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/conditional.m2.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/conditional.m2.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/conditional.m2.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/conditional.m2.qtl.out.RData
Untracked: output/conditional_m2.Novelty_residuals_RankNormal_datarelease_07302918_genomescan.pdf
Untracked: output/conditional_m2.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/genotype_error_marker.pdf
Untracked: output/genotype_frequency_marker.pdf
Untracked: output/m1.Novelty_resids_datarelease_07302918.RData
Untracked: output/m1.Novelty_resids_datarelease_07302918_coeffgeneplot.pdf
Untracked: output/m1.Novelty_resids_datarelease_07302918_genomescan.pdf
Untracked: output/m1.Novelty_resids_datarelease_12182019.RData
Untracked: output/m1.Novelty_resids_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m1.Novelty_resids_datarelease_12182019_genomescan.pdf
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_07302918_genomescan.pdf
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m1.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/m2.69k.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/m2.69k.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m2.69k.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/m2.69k.Prj01_RL-Acquisition_preqc_02142020.RData
Untracked: output/m2.69k.Prj01_RL-Reversal_preqc_02142020.RData
Untracked: output/m2.69k.Prj02_Sensitization_preqc_02142020.RData
Untracked: output/m2.69k.Prj04_IVSA_preqc_02142020.RData
Untracked: output/m2.Novelty_resids_datarelease_07302918.RData
Untracked: output/m2.Novelty_resids_datarelease_07302918_genomescan.pdf
Untracked: output/m2.Novelty_resids_datarelease_12182019.RData
Untracked: output/m2.Novelty_resids_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m2.Novelty_resids_datarelease_12182019_genomescan.pdf
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_07302918_genomescan.pdf
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m2.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/m2.Prj01_RL-Acquisition_preqc_02142020.RData
Untracked: output/m2.Prj01_RL-Acquisition_preqc_02142020_qtl2scan.pdf
Untracked: output/m2.Prj01_RL-Reversal_preqc_02142020.RData
Untracked: output/m2.Prj01_RL-Reversal_preqc_02142020_qtl2scan.pdf
Untracked: output/m2.Prj02_Sensitization_preqc_02142020.RData
Untracked: output/m2.Prj02_Sensitization_preqc_02142020_qtl2scan.pdf
Untracked: output/m2.Prj04_IVSA_preqc_02142020.RData
Untracked: output/m2.Prj04_IVSA_preqc_02142020_qtl2scan.pdf
Untracked: output/m3.Novelty_resids_datarelease_07302918.RData
Untracked: output/m3.Novelty_resids_datarelease_07302918_genomescan.pdf
Untracked: output/m3.Novelty_resids_datarelease_12182019.RData
Untracked: output/m3.Novelty_resids_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m3.Novelty_resids_datarelease_12182019_genomescan.pdf
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_07302918.RData
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_07302918_genomescan.pdf
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_12182019.RData
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_12182019_coeffgeneplot.pdf
Untracked: output/m3.Novelty_residuals_RankNormal_datarelease_12182019_genomescan.pdf
Untracked: output/num.geno.pheno.in.Novelty_resids_datarelease_07302918.csv
Untracked: output/num.geno.pheno.in.Novelty_resids_datarelease_12182019.csv
Untracked: output/num.geno.pheno.in.Novelty_residuals_RankNormal_datarelease_07302918.csv
Untracked: output/num.geno.pheno.in.Novelty_residuals_RankNormal_datarelease_12182019.csv
Untracked: output/number_crossover.pdf
Untracked: output/permu/
Untracked: output/prop_across_generation_chr_p.RData
Unstaged changes:
Modified: README.md
Modified: _workflowr.yml
Deleted: analysis/01_geneseek2qtl2.R
Deleted: analysis/02_geneseek2intensity.R
Deleted: analysis/03_firstgm2genoprobs.R
Deleted: analysis/04_diagnosis_qc_gigamuga_11_batches.Rmd
Deleted: analysis/04_diagnosis_qc_gigamuga_nine_batches.R
Deleted: analysis/04_diagnosis_qc_gigamuga_nine_batches.Rmd
Deleted: analysis/05_after_diagnosis_qc_gigamuga_11_batches.Rmd
Deleted: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.R
Deleted: analysis/05_after_diagnosis_qc_gigamuga_nine_batches.Rmd
Deleted: analysis/06_final_pr_apr_69K.R
Deleted: analysis/07.1_html_founder_prop.R
Deleted: analysis/07_recomb_size_founder_prop.R
Deleted: analysis/07_recomb_size_founder_prop.Rmd
Deleted: analysis/08_gcta_herit.R
Deleted: analysis/09_qtlmapping.R
Deleted: analysis/10_qtl_permu.R
Deleted: analysis/11_qtl_blup.R
Deleted: analysis/12_plot_69k_qtl_mapping_2.Rmd
Deleted: analysis/12_plot_qtl_mapping_1.Rmd
Deleted: analysis/12_plot_qtl_mapping_2.Rmd
Deleted: analysis/13_plot_69k_conditional_m2_qtlmapping.Rmd
Deleted: analysis/13_plot_conditional_m2_qtlmapping.Rmd
Deleted: analysis/16_diagnosis_qc_gigamuga_gagnon.Rmd
Deleted: analysis/16_diagnosis_qc_gigamuga_gagnon2.Rmd
Deleted: analysis/17_plot_qtl_mapping.Rmd
Deleted: analysis/run_01_geneseek2qtl2.R
Deleted: analysis/run_02_geneseek2intensity.R
Deleted: analysis/run_03_firstgm2genoprobs.R
Deleted: analysis/run_04_diagnosis_qc_gigamuga_nine_batches.R
Deleted: analysis/run_05_after_diagnosis_qc_gigamuga_nine_batches.R
Deleted: analysis/run_06_final_pr_apr_69K.R
Deleted: analysis/run_07.1_html_founder_prop.R
Deleted: analysis/run_07_recomb_size_founder_prop.R
Deleted: analysis/run_08_gcta_herit.R
Deleted: analysis/run_09_qtlmapping.R
Deleted: analysis/run_10_qtl_permu.R
Deleted: analysis/run_11_qtl_blup.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/05_after_diagnosis_qc_gigamuga_12_batches.Rmd) and HTML (docs/05_after_diagnosis_qc_gigamuga_12_batches.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 6610ab1 | xhyuo | 2020-11-05 | Build site. |
| Rmd | 8d3b7b7 | xhyuo | 2020-11-04 | 12_batches_before_afterQC |
| html | 070d088 | xhyuo | 2020-11-04 | Build site. |
| Rmd | 1b61cb6 | xhyuo | 2020-11-04 | 12_batches_before_afterQC |
After genotype diagnostics for diversity outbred mice
We first load the R/qtl2 package and the data. We’ll also load the R/broman package for some utilities and plotting functions, and R/qtlcharts for interactive graphs.
library
library(broman)
library(qtl2)
library(qtlcharts)
library(ggplot2)
library(ggrepel)
#library(DOQTL)
library(mclust)
library(tidyverse)
library(reshape2)
library(DT)
source("code/reconst_utils.R")
options(stringsAsFactors = F)Missing data per sample
gm <- get(load("data/Jackson_Lab_12_batches/gm_DO3173_qc.RData"))
gmObject of class cross2 (crosstype "do")
Total individuals 3173
No. genotyped individuals 3173
No. phenotyped individuals 3173
No. with both geno & pheno 3173
No. phenotypes 1
No. covariates 5
No. phenotype covariates 0
No. chromosomes 20
Total markers 112470
No. markers by chr:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
8532 8649 6402 6603 6558 6429 6281 5660 5856 5438 6339 5151 5259 5027 4548 4356
17 18 19 X
4322 3986 3103 3971 percent_missing <- n_missing(gm, "ind", "prop")*100
setScreenSize(height=100, width=300)Set screen size to height=100 x width=300labels <- paste0(names(percent_missing), " (", round(percent_missing,2), "%)")
iplot(seq_along(percent_missing), percent_missing, indID=labels,
chartOpts=list(xlab="Mouse", ylab="Percent missing genotype data",
ylim=c(0, 60)))#save into pdf
pdf(file = "data/Jackson_Lab_12_batches/AfterQC_Percent_missing_genotype_data.pdf", width = 20, height = 20)
labels <- as.character(do.call(rbind.data.frame, strsplit(ind_ids(gm), "V01_"))[,2])
labels[percent_missing < 5] = ""
# Change point shapes and colors
p <- ggplot(data = data.frame(Mouse=seq_along(percent_missing),
Percent_missing_genotype_data = percent_missing,
batch = factor(as.character(do.call(rbind.data.frame, strsplit(ind_ids(gm), "_"))[,5]))),
aes(x=Mouse, y=Percent_missing_genotype_data, color = batch)) +
geom_point() +
geom_hline(yintercept=5, linetype="solid", color = "red") +
geom_text_repel(aes(label=labels), vjust = 0, nudge_y = 0.01, show.legend = FALSE, size=3) +
theme(text = element_text(size = 20))
p
dev.off()png
2 p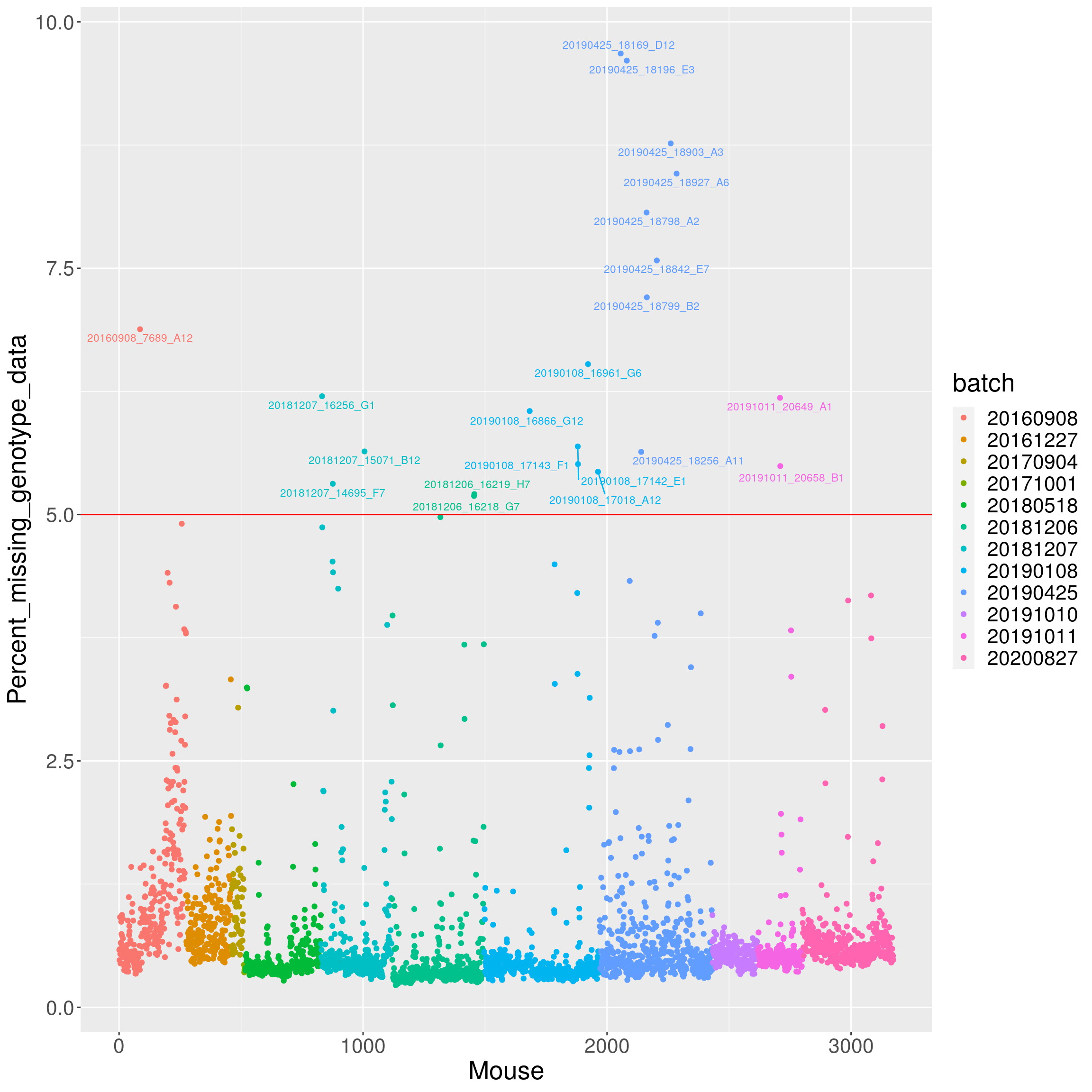
| Version | Author | Date |
|---|---|---|
| 070d088 | xhyuo | 2020-11-04 |
Sexes
xint <- read_csv_numer("data/Jackson_Lab_12_batches/Jackson_Lab_12_batches_qtl2_chrXint.csv", transpose=TRUE)
yint <- read_csv_numer("data/Jackson_Lab_12_batches/Jackson_Lab_12_batches_qtl2_chrYint.csv", transpose=TRUE)
#subset to gm subject name
xint <- xint[rownames(xint) %in% ind_ids(gm),]
yint <- yint[rownames(yint) %in% ind_ids(gm),]
# Gigamuga marker annotation file from UNC.
gm_marker_file = "http://csbio.unc.edu/MUGA/snps.gigamuga.Rdata" #FIXED
# Read in the UNC GigaMUGA SNPs and clusters.
load(url(gm_marker_file))
#subset down to gm
snps$marker = as.character(snps$marker)
#load the intensities.fst.RData
load("data/Jackson_Lab_12_batches/intensities.fst.RData")
#X and Y channel
X <- result[result$channel == "X",]
rownames(X) <- X$snp
X <- X[,c(-1,-2)]
Y <- result[result$channel == "Y",]
rownames(Y) <- Y$snp
Y <- Y[,c(-1,-2)]
#determine predict.sex
predict.sex = determine_sex_chry_m(x = X, y = Y, markers = snps)$sex
gm$covar <- gm$covar %>%
mutate(id = rownames(gm$covar)) %>%
left_join(data.frame(id = names(predict.sex),
predict.sex = predict.sex,stringsAsFactors = F))Joining, by = c("id", "predict.sex")rownames(gm$covar) <- gm$covar$id
#sex order
sex <- gm$covar[rownames(xint),"sex"]
x_pval <- apply(xint, 2, function(a) t.test(a ~ sex)$p.value)
y_pval <- apply(yint, 2, function(a) t.test(a ~ sex)$p.value)
xint_ave <- rowMeans(xint[, x_pval < 0.05/length(x_pval)], na.rm=TRUE)
yint_ave <- rowMeans(yint[, y_pval < 0.05/length(y_pval)], na.rm=TRUE)
point_colors <- as.character( brocolors("web")[c("green", "purple")] )
labels <- paste0(names(xint_ave))
iplot(xint_ave, yint_ave, group=sex, indID=labels,
chartOpts=list(pointcolor=point_colors, pointsize=4,
xlab="Average X chr intensity", ylab="Average Y chr intensity"))phetX <- rowSums(gm$geno$X == 2)/rowSums(gm$geno$X != 0)
iplot(xint_ave, phetX, group=sex, indID=labels,
chartOpts=list(pointcolor=point_colors, pointsize=4,
xlab="Average X chr intensity", ylab="Proportion het on X chr"))Sample duplicates
cg <- compare_geno(gm, cores=10)
summary.cg <- summary(cg, threshold = 0.65)
#get the name and missing percentage
summary.cg$Name.ind1 <- str_split_fixed(summary.cg$ind1, "_",7)[,6]
summary.cg$Name.ind2 <- str_split_fixed(summary.cg$ind2, "_",7)[,6]
summary.cg$miss.ind1 <- percent_missing[match(summary.cg$ind1, names(percent_missing))]
summary.cg$miss.ind2 <- percent_missing[match(summary.cg$ind2, names(percent_missing))]
summary.cg$remove.id <- ifelse(summary.cg$miss.ind1 > summary.cg$miss.ind2, summary.cg$ind1, summary.cg$ind2)
#display summary.cg
DT::datatable(summary.cg, filter = list(position = 'top', clear = FALSE),
options = list(pageLength = 40, scrollY = "300px", scrollX = "40px"))pdf(file = "data/Jackson_Lab_12_batches/AfterQC_Proportion_matching_genotypes_before_removal_of_bad_samples.pdf", width = 20, height = 20)
par(mar=c(5.1,0.6,0.6, 0.6))
hist(cg[upper.tri(cg)], breaks=seq(0, 1, length=201),
main="", yaxt="n", ylab="", xlab="Proportion matching genotypes")
rug(cg[upper.tri(cg)])
dev.off()png
2 par(mar=c(5.1,0.6,0.6, 0.6))
hist(cg[upper.tri(cg)], breaks=seq(0, 1, length=201),
main="", yaxt="n", ylab="", xlab="Proportion matching genotypes")
rug(cg[upper.tri(cg)])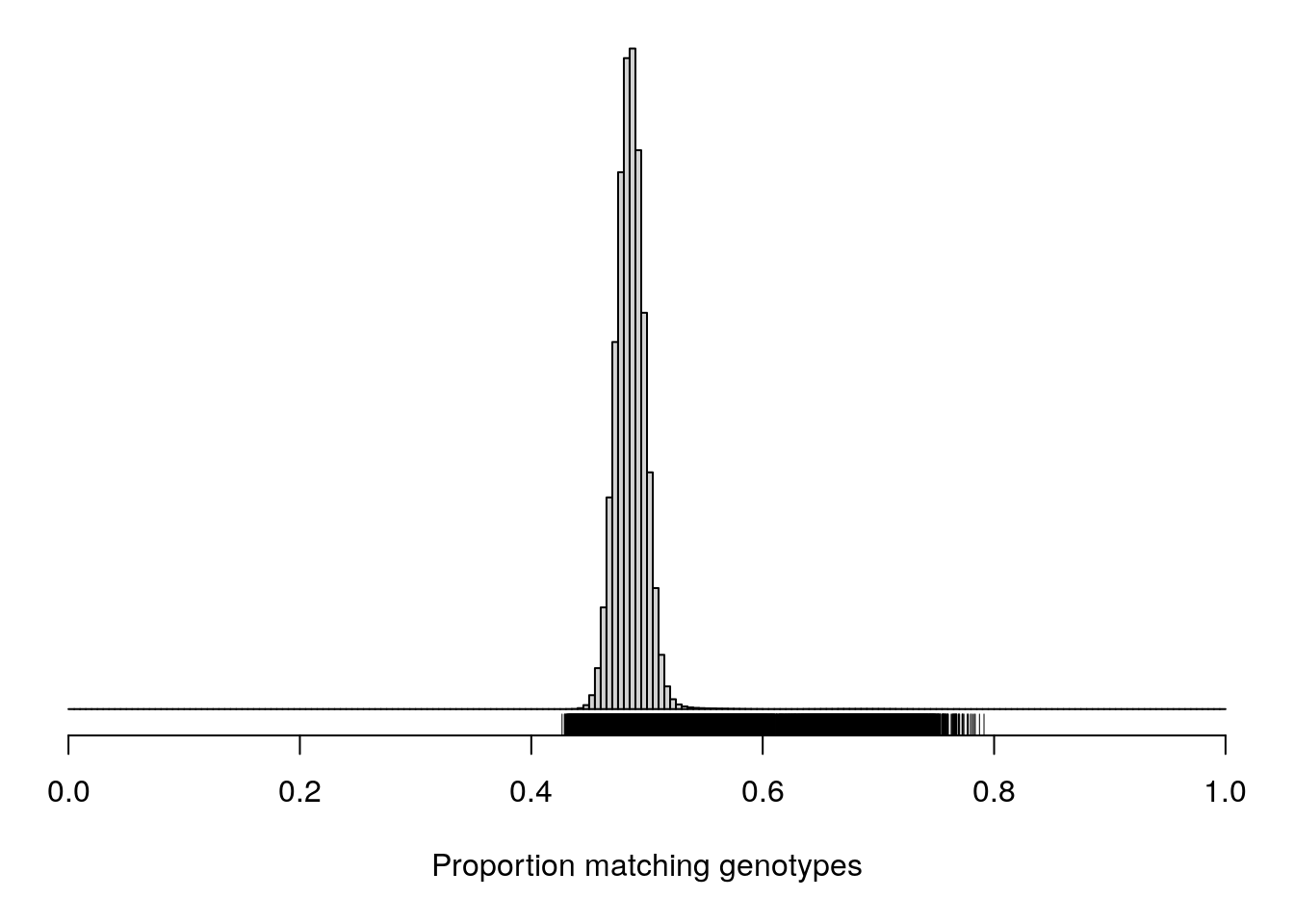
| Version | Author | Date |
|---|---|---|
| 070d088 | xhyuo | 2020-11-04 |
pdf(file = "data/Jackson_Lab_12_batches/AfterQC_Proportion_matching_genotypes_after_removal_of_bad_samples.pdf",width = 20, height = 20)
cgsub <- cg[percent_missing < 5, percent_missing < 5]
par(mar=c(5.1,0.6,0.6, 0.6))
hist(cgsub[upper.tri(cgsub)], breaks=seq(0, 1, length=201),
main="", yaxt="n", ylab="", xlab="Proportion matching genotypes")
rug(cgsub[upper.tri(cgsub)])
dev.off()png
2 cgsub <- cg[percent_missing < 5, percent_missing < 5]
par(mar=c(5.1,0.6,0.6, 0.6))
hist(cgsub[upper.tri(cgsub)], breaks=seq(0, 1, length=201),
main="", yaxt="n", ylab="", xlab="Proportion matching genotypes")
rug(cgsub[upper.tri(cgsub)])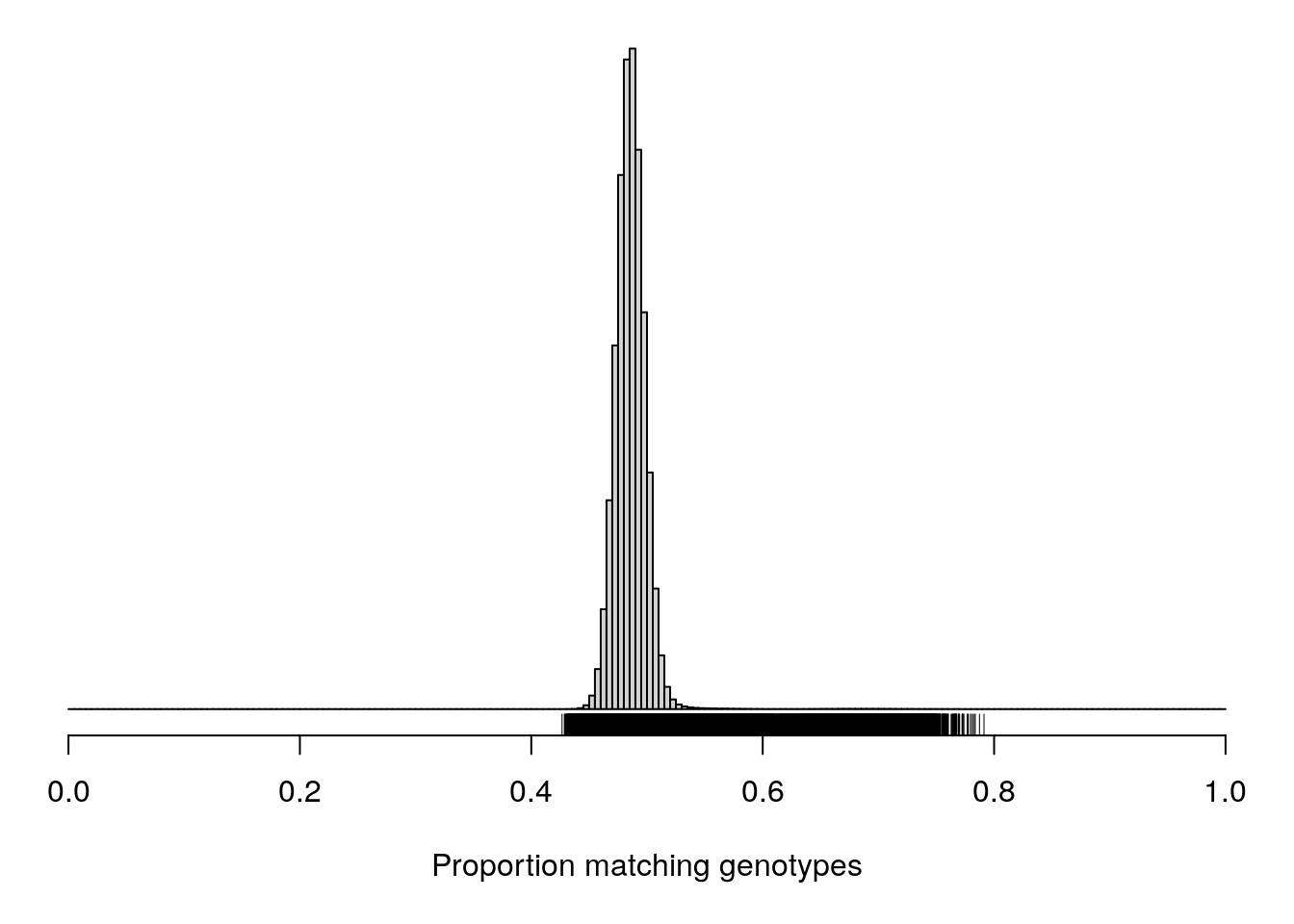
| Version | Author | Date |
|---|---|---|
| 070d088 | xhyuo | 2020-11-04 |
#show top 20 samples with missing genotypes
percent_missing <- n_missing(gm, "ind", "prop")*100
round(sort(percent_missing, decreasing=TRUE)[1:20], 1)Jackson_Lab_Bubier_MURGIGV01_20190425_18169_D12
9.7
Jackson_Lab_Bubier_MURGIGV01_20190425_18196_E3
9.6
Jackson_Lab_Bubier_MURGIGV01_20190425_18903_A3
8.8
Jackson_Lab_Bubier_MURGIGV01_20190425_18927_A6
8.5
Jackson_Lab_Bubier_MURGIGV01_20190425_18798_A2
8.1
Jackson_Lab_Bubier_MURGIGV01_20190425_18842_E7
7.6
Jackson_Lab_Bubier_MURGIGV01_20190425_18799_B2
7.2
Jackson_Lab_Bubier_MURGIGV01_20160908_7689_A12
6.9
Jackson_Lab_Bubier_MURGIGV01_20190108_16961_G6
6.5
Jackson_Lab_Bubier_MURGIGV01_20181207_16256_G1
6.2
Jackson_Lab_Gagnon_MURGIGV01_20191011_20649_A1
6.2
Jackson_Lab_Bubier_MURGIGV01_20190108_16866_G12
6.1
Jackson_Lab_Bubier_MURGIGV01_20190108_17142_E1
5.7
Jackson_Lab_Bubier_MURGIGV01_20181207_15071_B12
5.6
Jackson_Lab_Bubier_MURGIGV01_20190425_18256_A11
5.6
Jackson_Lab_Bubier_MURGIGV01_20190108_17143_F1
5.5
Jackson_Lab_Gagnon_MURGIGV01_20191011_20658_B1
5.5
Jackson_Lab_Bubier_MURGIGV01_20190108_17018_A12
5.4
Jackson_Lab_Bubier_MURGIGV01_20181207_14695_F7
5.3
Jackson_Lab_Bubier_MURGIGV01_20181206_16219_H7
5.2 Array intensities and Genotype frequencies
int <- result[,c("snp","channel",ind_ids(gm))]
#rm(result)
int <- int[seq(1, nrow(int), by=2),-(1:2)] + int[-seq(1, nrow(int), by=2),-(1:2)]
int <- int[,intersect(ind_ids(gm), colnames(int))]
n <- names(sort(percent_missing[intersect(ind_ids(gm), colnames(int))], decreasing=TRUE))
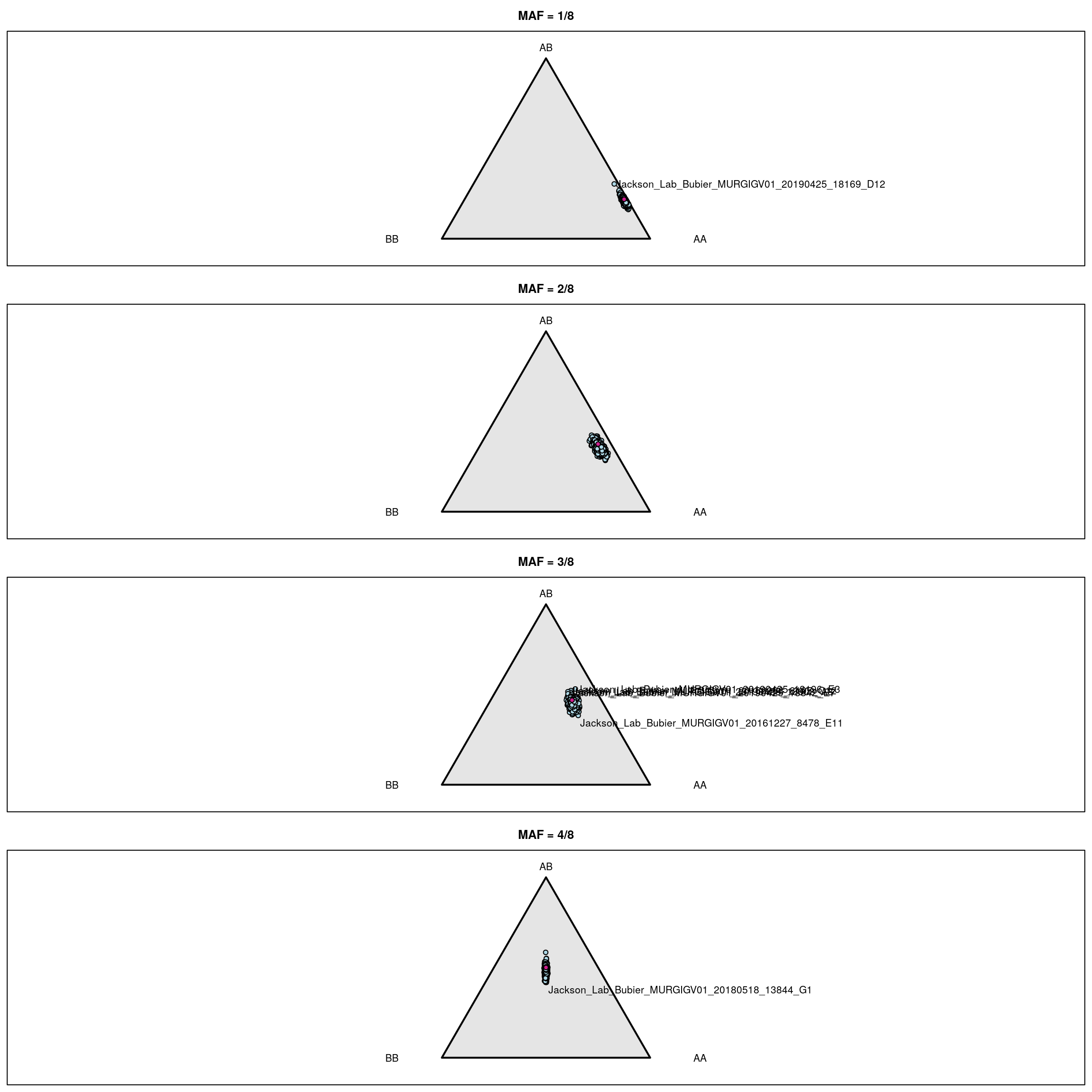
iboxplot(log10(t(int[,n])+1), orderByMedian=FALSE, chartOpts=list(ylab="log10(SNP intensity + 1)"))# Genotype frequencies
g <- do.call("cbind", gm$geno[1:19])
fg <- do.call("cbind", gm$founder_geno[1:19])
g <- g[,colSums(fg==0)==0]
fg <- fg[,colSums(fg==0)==0]
fgn <- colSums(fg==3)
gf_ind <- vector("list", 4)
for(i in 1:4) {
gf_ind[[i]] <- t(apply(g[,fgn==i], 1, function(a) table(factor(a, 1:3))/sum(a != 0)))
}
par(mfrow=c(4,1), mar=c(0.6, 0.6, 2.6, 0.6))
for(i in 1:4) {
triplot(c("AA", "AB", "BB"), main=paste0("MAF = ", i, "/8"))
tripoints(gf_ind[[i]], pch=21, bg="lightblue")
tripoints(c((1-i/8)^2, 2*i/8*(1-i/8), (i/8)^2), pch=21, bg="violetred")
if(i>=3) { # label mouse with lowest het
wh <- which(gf_ind[[i]][,2] == min(gf_ind[[i]][,2]))
tritext(gf_ind[[i]][wh,,drop=FALSE] + c(0.02, -0.02, 0),
names(wh), adj=c(0, 1))
}
# label other mice
if(i==1) {
lab <- rownames(gf_ind[[i]])[gf_ind[[i]][,2]>0.3]
}
else if(i==2) {
lab <- rownames(gf_ind[[i]])[gf_ind[[i]][,2]>0.48]
}
else if(i==3) {
lab <- rownames(gf_ind[[i]])[gf_ind[[i]][,2]>0.51]
}
else if(i==4) {
lab <- rownames(gf_ind[[i]])[gf_ind[[i]][,2]>0.6]
}
for(ind in lab) {
if(grepl("^F", ind) && i != 3) {
tritext(gf_ind[[i]][ind,,drop=FALSE] + c(-0.01, 0, +0.01), ind, adj=c(1,0.5))
} else {
tritext(gf_ind[[i]][ind,,drop=FALSE] + c(0.01, 0, -0.01), ind, adj=c(0,0.5))
}
}
}
| Version | Author | Date |
|---|---|---|
| 070d088 | xhyuo | 2020-11-04 |
Crossover counts and Genotyping error LOD scores
#load pre-caluated results
load("data/Jackson_Lab_12_batches/nxo.RData")
#crossover
totxo <- rowSums(nxo)[names(rowSums(nxo)) %in% ind_ids(gm)]
totxo <- totxo[ind_ids(gm)]
all.equal(ind_ids(gm), names(totxo))[1] TRUEiplot(seq_along(totxo),
totxo,
group=gm$covar$ngen,
chartOpts=list(xlab="Mouse", ylab="Number of crossovers",
margin=list(left=80,top=40,right=40,bottom=40,inner=5),
axispos=list(xtitle=25,ytitle=50,xlabel=5,ylabel=5)))#save crossover into pdf
pdf(file = "data/Jackson_Lab_12_batches/AfterQC_number_crossover.pdf")
cross_over <- data.frame(Mouse = seq_along(totxo), Number_crossovers = totxo, generation = gm$covar$ngen)
names(totxo) <- as.character(do.call(rbind.data.frame, strsplit(names(totxo), "V01_"))[,2])
names(totxo)[totxo <= 800] = ""
# Change point shapes and colors
p <-ggplot(cross_over, aes(x=Mouse, y=Number_crossovers, fill = generation, color=generation)) +
geom_point() +
geom_text_repel(aes(label=names(totxo),hjust=0,vjust=0), show.legend = FALSE)
p
dev.off()png
2 p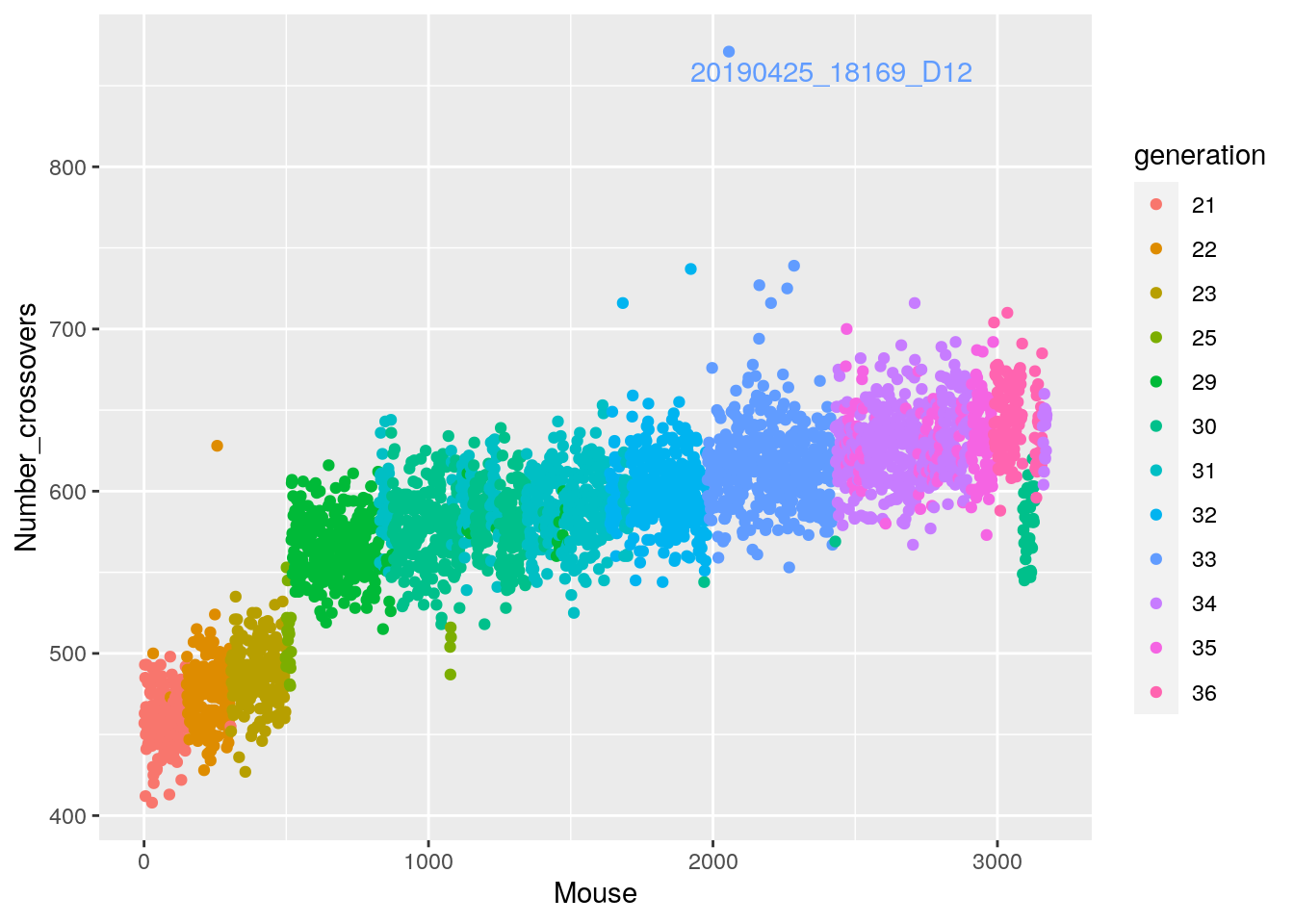
| Version | Author | Date |
|---|---|---|
| 070d088 | xhyuo | 2020-11-04 |
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.4 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] DT_0.13 reshape2_1.4.4 forcats_0.5.0 stringr_1.4.0
[5] dplyr_1.0.0 purrr_0.3.4 readr_1.4.0 tidyr_1.1.0
[9] tibble_3.0.1 tidyverse_1.3.0 mclust_5.4.6 ggrepel_0.8.2
[13] ggplot2_3.3.2 qtlcharts_0.12-10 qtl2_0.22-8 broman_0.70-4
[17] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.4.6 lubridate_1.7.9 assertthat_0.2.1 rprojroot_1.3-2
[5] digest_0.6.25 plyr_1.8.6 R6_2.4.1 cellranger_1.1.0
[9] backports_1.1.6 reprex_0.3.0 RSQLite_2.2.0 evaluate_0.14
[13] httr_1.4.1 pillar_1.4.4 rlang_0.4.6 readxl_1.3.1
[17] rstudioapi_0.11 data.table_1.12.8 whisker_0.4 blob_1.2.1
[21] rmarkdown_2.5 labeling_0.4.2 qtl_1.46-2 htmlwidgets_1.5.1
[25] bit_1.1-15.2 munsell_0.5.0 broom_0.7.2 compiler_4.0.0
[29] httpuv_1.5.4 modelr_0.1.8 xfun_0.13 pkgconfig_2.0.3
[33] htmltools_0.4.0 tidyselect_1.1.0 fansi_0.4.1 crayon_1.3.4
[37] dbplyr_2.0.0 withr_2.2.0 later_1.0.0 grid_4.0.0
[41] jsonlite_1.6.1 gtable_0.3.0 lifecycle_0.2.0 DBI_1.1.0
[45] git2r_0.27.1 magrittr_1.5 scales_1.1.1 cli_2.0.2
[49] stringi_1.4.6 farver_2.0.3 fs_1.4.1 promises_1.1.0
[53] xml2_1.3.2 ellipsis_0.3.0 generics_0.0.2 vctrs_0.3.1
[57] tools_4.0.0 bit64_0.9-7 glue_1.4.0 crosstalk_1.1.0.1
[61] hms_0.5.3 parallel_4.0.0 yaml_2.2.1 colorspace_1.4-1
[65] rvest_0.3.6 memoise_1.1.0 knitr_1.28 haven_2.3.1