Analysing 16S data: Part 1
Session Overview
During this session we will cover the fundamentals of amplicon-based microbiome analysis.
Details of the individual session components are included below:
1. Using a text editor to document your actions
2. An introduction to sequence data
3. Combining paired-end reads into contiguous sequences
4. Generating a unique set of 16S gene sequences
5. Clustering unique sequences into operational taxonomic units
6. Creating a table of OTU abundance
7. Assigning taxonomy to OTU sequences
Session One
1. Using a text editor to document your actions
During this session you may wish to keep a record of the commands used to analyse your 16S data. One way to do this is to write each command to a file. From the command line, this can be acheived using a text editor. There are many to choose from, but this session will use Nano.
# Navigate to the working directory for this session
cd ~/MCA/16s/Session1
# Open a new file
nano 16s_pipeline.shOpening a new text file you should see something similar to this:
Try writing some text to the document 16s_pipeline.sh
Try saving the document, closing it, and re-opening it.
Because the saved document is a shell script, it is also possible to run the entire script from the command line.
# Make the file 16s_pipeline.sh executable from the command line.
chmod u+x 16s_pipeline.sh
# run the code in 16s_pipeline.sh
./16s_pipeline.shFor more information on using nano, see the Nano online documentation.
2. An introduction to 16S sequence data
The ubiquitous format for the storage of sequence data are fastq files. To begin, navigate to the location of the sequence data that will be used for today’s session.
cd ~/MCA/16s/Session1/fastqs/ # change directory
ls # list contents of directoryThese data are from an Illumina paired-end sequencing run. There should be two files per sample, with the files *.R1_sub.fastq and *.R2_sub.fastq containing the first and second reads in each pair, respectively. Have a look at the first four lines in one of the fastqs.
head -4 A_control.R1_sub.fastq # view the first n lines of a file
@M03204:217:000000000-B8W8J:1:2108:24791:7353
AGAGTTTGATCCTGGCTCAGGATGAACGCTGGCGGCATGCCTTACACATGCAAGTCGGACGGGAAGTGGTGTTTCCAGTGGCGGACGGGTGAGTAACGCGTAAGAACCTACCCTTGGGAGGGGAACAACAGCTGGAAACGGCTGCTAATACCCCGTAGGCTGAGGAGCAAAAGGAGGAATCCGCCCGAGGAGGGGTTCGCGTCTGATTAGCTAGTTGGTGAGGCAATAGCTTACCAAGGCGATGATCAGTAGCTGGTCCGAGAGGATGATCAGCCACACTGGGACTGAGACACGGCCCAGA
+
CCCCCGFGGGGGGFCFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDDFGGGGGGGGGGGGGGGGGGGFGGGGDGGGGGGGGGFGGGGGGEGB:FEFGGGFGGECGGGGFFGFGFGGGGGGGGFGFEGGGGGGFFFGGGGGGFECGGGFGGGGGGGGGGGGGGGGGGGGF6CFFGGGGDGGEFFCF:5*)5>29*5<)5>GF4)What information is contained in each line of the fastq?
For further detail on the contents of fastq files, see here.
3. Combining paired-end reads into contiguous sequences
In order to generate a single, contiguous sequence spanning the target region of the 16S gene, it’s necessary the paired reads in each fastq file. To do this we will use a tool called FLASh. Start by combining two fastq files for a single sample.
# Check that you have returned to the main session directory
# This should be ~/MCA/16s/Session1
pwd
# Run FLASh for a single sample
flash ./fastqs/A_control.R1_sub.fastq ./fastqs/A_control.R2_sub.fastq
# The files created by FLASh should be in the current directory
lsHave a look at the files created by FLASh, in particular the file out.extendedFrags.fastq
Are all sequences the same length?
For information on options for running FLASh and the files it creates visit the website, or access the help documentation on the command line.
flash --help3.b. Running FLASh using a for loop
While it’s possible to run FLASh individually for each sample. It’s also possible to use a for loop to iteratively combine fastqs for all the samples in the fastq directory.
# make a directory in which to store the combined fastq files
mkdir fastqs_combined
ls
ls fastqs_combined
# Run FLASh for all samples in the working directory
for FASTQ1 in fastqs/*.R1_sub.fastq
do
# capture the file prefix, which corresponds to the sample name
SAMPLEID="$(basename ${FASTQ1%.R1_sub.fastq})"
# assign the second fastq file to the variable FASTQ2
FASTQ2="fastqs/${SAMPLEID}.R2_sub.fastq"
# run FLASh
flash $FASTQ1 $FASTQ2 --output-prefix=$SAMPLEID --output-directory=fastqs_combined
done
ls fastqs_combinedUsing your text editor, copy the for loop into your pipeline script.
Remember to annotate your code so that you can return to it later.
3.c. Converting fastq files to fasta format
From here on, it’s necessary to work with fasta files, rather than fastq. It’s possible to convert our data from fastq to fasta format using the FASTX-toolkit.
# Run fastq_to_fasta for the combined fastq files created by FLASh
for FASTQ in fastqs_combined/*.extendedFrags.fastq
do
# capture the file prefix, which corresponds to the sample name
SAMPLEID="${FASTQ%.extendedFrags.fastq}"
# run fastq_to_fasta
fastq_to_fasta -i $FASTQ -o $SAMPLEID.fasta
done
# The fasta files should now be in the same directory as the combined fastqs
ls fastqs_combinedEnter the combined_fastq directory and look at the first four lines of a fasta file. How do they differ from the fastq format?
4. Generating a unique set of 16S gene sequences
Having successfully processed our sequence data, the goals now are to:
- Identify distinct biological taxa
- Quantify the relative abundance of each distinct taxon
These goals are predicated on the assumption that the extent to which two distinct bacterial taxa are related correlates with the similarity of their 16S rRNA gene sequences. Different taxa can therefore be identified by resolving unique 16S gene sequences and counting their abundance.
There are many ways to acheive these goals. However, in this session we will make use of the widely used tool USEARCH, which can accomplish all of these steps.
The first step in processing data for use with USEARCH is to pool 16S gene sequences from different samples into one file. In order to keep track of which sequence originated from which sample, it is also necessary to add the sample name to the header line for each fasta sequence. For simplicity, this step will be carried out using a custom script.
# The location of the script for combining fasta files
which samples2fasta
# Combine samples into a single file located in the run directory
samples2fasta fastqs_combined all_samples.fastaThe second step is to create a file containing a single copy of each unique sequence in the
dataset, as each unique sequence potentially represents a unique bacterial taxon. When the
-sizeout argument is provided USEARCH keeps a record of the number of times each unique
sequence appears in the data set.
# How many sequences are in the combined fasta file?
cat all_samples.fasta | grep ">" | wc -l
# Run USEARCH to generate a file of unique sequences
usearch -fastx_uniques all_samples.fasta -fastaout all_unique_seqs.fasta -sizeoutHow many unique sequences remain after removing duplicate copies? Have a look at the first four lines of
all_unique_seqs.fastato see where sizeout is recorded.
Sequence variation may be a result of divergent evolution, but it may also be caused by errors during sequencing. If a bacterial taxon is present at a detectable abundance in these samples, then its 16S gene sequence is likely to be represented multiple times in the data set. By contrast, sequencing error is assumed to be (more-or-less) random, meaning that errors due to sequencing have only a small likelihood of occurring more than once. For this reason it is common practice to discard unique sequences that occur one, or a few times. The next step is to sort unique sequences based on their frequency of occurrence, and to discard those that occur only once.
# Sort unique sequences based on the number of times they occur, discarding singletons
usearch -sortbysize all_unique_seqs.fasta -fastaout all_unique_seqs_sorted.fasta -minsize 2How many unique sequences remain after removing singletons?
How many unique sequences remain if you discard sequences occurring less than five times?
5. Clustering unique sequences into operational taxonomic units
As discussed in Section 4, sequence-based analysis assumes that taxonomic (or biologically relevant?) differences between bacteria are reflected by differences in their 16S gene sequence. But how much do two 16S gene sequences have to differ before they represent two distinct bacterial strains, species, or even genera? There is a discussion of this issue on the USEARCH website.
In this practical session the goal is to identify and quantify distinct bacterial species. Assuming that a certain amount of 16S gene sequence variation exists within species (different bacterial strains?) we will generate operational taxonomic units (OTUs) based on the assumption that two sequences which are more than 97% similar belong to the same species.
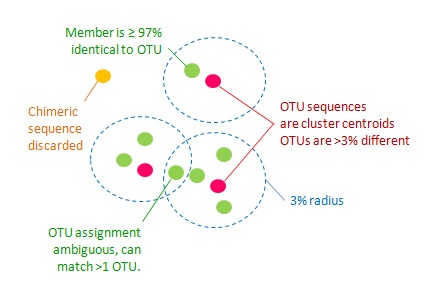
The UPARSE-OTU algorithm, implemented in USEARCH, can be used to generate OTUs based on this 97% similarity threshold. It selects a single representative sequence for each generated OTU.
# Cluster OTUs using a 97% similarity threshold
usearch -cluster_otus all_unique_seqs_sorted.fasta -otus otus.fastaHow many OTUs have been generated?
How many chimeras were detected?
How does the number of OTUs compare to the number of sequences in the combined fasta file produced in Section 3c?
Note that chimeras can be a significant problem in amplicon-based sequence analysis. There are dedicated tools for chimera detection and removal (e.g. ChimeraSlayer); however the UPARSE-OTU algorithm implicitly filters chimeras.
6. Creating a table of OTU abundance
Having generated a set of sequences representing distinct bacterial taxa (OTUs). The next step is to quantify OTU abundance. This can be acheived by counting the number of sequences in each sample that match each OTU. For a sequence to “match” an OTU it must be more than 97% similar to the representative sequence for that OTU.
The first step is to relabel each of our OTUs with a unique identifier. We will do this using a python script fasta_number.py
# location of the fasta_number.py python script
which fasta_number.py
# call python script to rename the OTUs in the format OTU_*
fasta_number.py otus.fasta OTU_ > otus_renamed.fastaThe next step is to use the pooled fasta file (all_samples.fasta) generated in
Section 4 to map the sequences originating from each sample file back their closest
matching OTUs. Sample sequences that do not match any OTU sequenced at >97% similarity are assumed
to be sequencing errors and are discarded.
# Match all sample sequences back to OTU sequences at a 97% similarity threshold
# Sample sequnences are present in the file all_samples.fasta
# The reference (-db) is the file containing representative OTU sequences
usearch -usearch_global \
all_samples.fasta \
-db otus_renamed.fasta \
-strand plus \
-id 0.97 \
-uc otu_map.ucHave a look at the format of the output file
otu_map.uc. Can you identify the information in each column?
Can you fine a line in this file that begins with “N”?
The output file generated when matching sample sequences to OTUs is in USEARCH cluster format. It contains the best OTU match (if any) for each sample sequence. From this file it is possible to generate a count table summarizing the number of sequences in each sample that match each OTU.
# find the location of the python script usearch2otutab.py
which uc2otutab.py
# call this script from it's installed location using python
python ~/local/bin/uc2otutab.py otu_map.uc > otu_matrix.tsvThe resulting file otu_matrix.tsv is a count matrix in which columns are samples, rows are OTUs
and each cell represents the number of sequences assigned to each OTU in each sample.
7. Assigning taxonomy to OTU sequences
Having generated a matrix of OTU abundance in different samples it is often useful to compare OTU sequences against a reference database in order to identify their likely taxonomy. One simple way to do this would be to copy each sequence into NCBI’s [BLAST][ncbi-ntblast] tool, which will compare it to the NCBI nucleotide database.
# Select the first OTU sequence in your renamed fasta
head -2 otus_renamed.fastaTry copying the entire OTU sequence into the “Enter Query Sequence” box on the NCBI blast
website. Then hit “BLAST”. How informative is the closest BLAST match in the NCBI nucleotide reference database?
An alternative method for classifying 16S gene sequences is to use the Ribosomal Database Project (RDP) classifier tool, which compares sequences to the RDP reference database. The RDP classifier can be run from the command line without the need to download the entire reference database.
# Run the RDP classifer. As this tool is written in java, it's necessary to first run
# java then pass the location of the installed classifier
java -Xmx1g -jar ~/local/src/rdp_classifier_2.12/dist/classifier.jar classify \
-f filterbyconf \
-c 0.8 \
-o otu_taxonomy_rdp_0.8.tsv \
-h otu_taxonomy.hierachy \
otus_renamed.fastaHave a look at the two files (
otu_taxonomy_rdp_0.8.tsvandotu_taxonomy.hierachy) produced by the RDP classifier.
The RDP classifier produces two files the first otu_taxonomy_rdp_0.8.tsv contains a taxonomic
classification from each OTU from Kingdom to Genus level. The second otu_taxonomy.hierachy
contains similar information in a commonly used hierachical format. Each taxonomic level encountered
in our dataset is listed in this file, with the final column showing the number of times it is
encountered.
For further information on the options available when running the RDP classifer have a look at the
help pages, as well as the README file that comes with the RDP installation.
# Running RDP without input arguments will cause it to print its help pages.
java -Xmx1g -jar ~/local/src/rdp_classifier_2.12/dist/classifier.jar
java -Xmx1g -jar ~/local/src/rdp_classifier_2.12/dist/classifier.jar classify
# For more detailed information look at the README file
less ~/local/src/rdp_classifier_2.12/READMEWhat does the command
-c 0.8mean?
Conclusions
This session provides an overview of the fundamental steps taken to process 16S gene sequence data from raw reads to a taxonomic abundance table that can be used for downstream analysis. It makes use of the freely available tools FLASh and USEARCH. However, there are many other excellent tools/SOPs available online. See for example Mothur, Qiime, and Tornado for further discussion of the issues surrounding 16S sequence analysis.
If you have managed to maintain your script 16s_pipeline.sh throughout this session, then you will
have a record of all the steps taken during 16S sequence processing. It should now be
possible to rerun this pipeline on this and other datasets to automatically go from
merging fastq files to generating a count matrix. Pipelines are an
essential part of high-throughput sequence analysis. For an interesting discussion of the importance
of reproducibility in modern biological research see here.