Running LIRICAL
LIRICAL is a command-line Java tool that runs with Java version 17 or higher. LIRICAL can be run both with and without genomic data in form of a VCF file from genome, exome, or NGS gene-panel sequencing.
On typical computers, LIRICAL will run from about 15 to 60 seconds in phenotype-only mode, ~5 minutes with a typical exome file, or longer if a whole-genome file is used as input.
To get help, run LIRICAL with a command or with the option -h:
lirical --help
LIkelihood Ratio Interpretation of Clinical AbnormaLities
Usage: lirical [-hV] [COMMAND]
-h, --help Show this help message and exit.
-V, --version Print version information and exit.
Commands:
download, D Download files for LIRICAL.
prioritize, R Run LIRICAL from CLI arguments.
phenopacket, P Run LIRICAL from a Phenopacket.
yaml, Y Run LIRICAL from a YAML file.
See the full documentation at https://lirical.readthedocs.io/en/master
Note
We assume that the lirical command alias was set as described in the Set up alias section.
Run LIRICAL with a specific command with the -h option to get information about the command:
lirical download -h
Usage: lirical download [-hVw] [-d=<datadir>]
Download files for LIRICAL.
-d, --data=<datadir> directory to download data (default: data)
-w, --overwrite overwrite previously downloaded files (default: false)
-h, --help Show this help message and exit.
-V, --version Print version information and exit.
LIRICAL has four main commands, download, prioritize, phenopacket, and yaml.
We will not discuss the download command since it has already been covered in the LIRICAL data files section
LIRICAL prioritization commands
LIRICAL provides three commands for receiving phenotype and genotype inputs via CLI, as a phenopacket, or as a YAML file.
prioritize - run LIRICAL with via CLI options
Since v2 release, all required inputs can be provided as command line arguments of the prioritize command.
This leads to a rather lengthy CLI. However, the CLI can be useful e.g. for using with pipeline engines such
as Nextflow or Snakemake.
The prioritize command takes the following options:
-p | --observed-phenotypes: a comma-separated IDs of HPO IDs that correspond to the phenotype terms observed in the proband.-n | --negated-phenotypes: a comma-separated IDs of HPO IDs that correspond to the phenotype terms negated/excluded in the proband.--assemblygenome build, choose from hg19 or hg38. The assembly must be provided if--vcfis used. If both--assemblyand--vcfare unset, the analysis will be run in phenotype-only mode.--vcf: path to VCF file with exome/genome sequencing results. The file can be compressed.--sample-id: proband’s identifier, must be provided if running with a multi-sample VCF file (default: subject).--age: proband’s age as an ISO8601 duration. (e.g.P9Yfor 9 years,P2Y3Mfor 2 years and 3 months, orP33Wfor the 33th gestational week).--sex: proband’s sex, choose from MALE, FEMALE, UNKNOWN (default: UNKNOWN).
phenopacket - run LIRICAL with a Phenopacket
LIRICAL can be run with clinical data (HPO terms) only or with clinical data and a VCF file representing the results of gene panel, exome, or genome sequencing. The preferred input format is Phenopackets, an open standard for sharing disease and phenotype information. This is a new standard of the Global Alliance for Genomics and Health that links detailed phenotype descriptions with disease, patient, and genetic information.
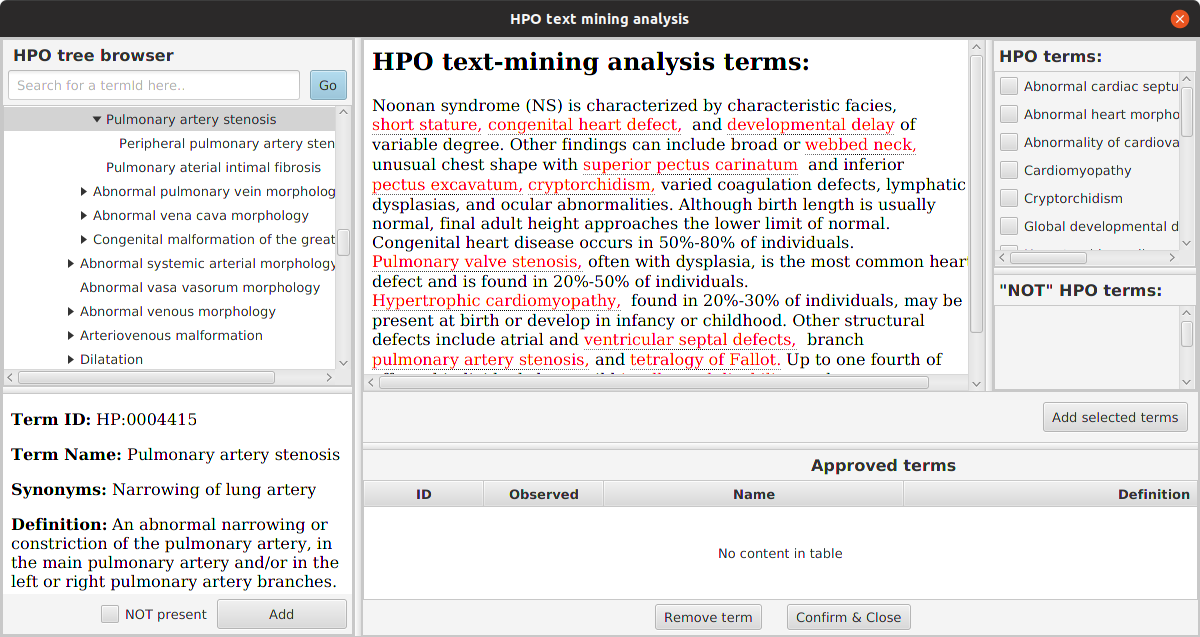
For convenience, we provide a tool called PhenopacketGenerator that can be used to create a Phenopacket with a list of HPO terms and the path to a VCF file with which LIRICAL can be run.
LIRICAL can be run with clinical data (HPO terms) only or with clinical data and a VCF file representing the results of gene panel, exome, or genome sequencing.
Let’s consider an example of an individual with Pfeiffer syndrome:
{
"id": "pfeiffer-example",
"subject": {
"id": "example-1"
},
"phenotypicFeatures": [{
"type": {
"id": "HP:0000244",
"label": "Turribrachycephaly"
}
}, {
"type": {
"id": "HP:0001363",
"label": "Craniosynostosis"
}
}, {
"type": {
"id": "HP:0000453",
"label": "Choanal atresia"
}
}, {
"type": {
"id": "HP:0000327",
"label": "Hypoplasia of the maxilla"
}
}, {
"type": {
"id": "HP:0000238",
"label": "Hydrocephalus"
}
}],
"metaData": {
"createdBy": "Peter R.",
"resources": [{
"id": "hp",
"name": "human phenotype ontology",
"namespacePrefix": "HP",
"url": "http://purl.obolibrary.org/obo/hp.owl",
"version": "2018-03-08",
"iriPrefix": "http://purl.obolibrary.org/obo/HP_"
}],
"phenopacketSchemaVersion": "2.0.0"
}
}
Save the file above as pfeiffer.json.
Running LIRICAL with clinical data
LIRICAL will perform phenotype-only analysis if the phenopacket command incantation does not contain
the --assembly and --vcf options.
In this case, the only required argument is the phenopacket:
lirical phenopacket -p pfeiffer.json
Running LIRICAL with a VCF file
Alternatively, LIRICAL can use a VCF file if the path is provided using --vcf option.
Note, the --assembly and -ed19 (or -ed38) options to indicate the genome assembly
and path to Exomiser data directory must also be provided:
lirical phenopacket \
--assembly hg19 \
--vcf path/to/pfeiffer.vcf.gz \
-ed19 /path/to/exomiser/datadir \
-p pfeiffer.json
yaml - running LIRICAL with a YAML file
The other allowed input format is YAML input format.
A typical command that runs LIRICAL using settings shown in the YAML file with the default data directory would be simply:
lirical yaml -y example.yml
This will run the phenotype-only analysis of the Patient 4.
To run the genotype-aware analysis, modify the YAML file such that the vcf field points to the location
of the VCF file on your file system. Then, the analysis is run as:
lirical yaml -y example.yml --assembly hg19 -ed19 /path/to/exomiser/datadir
Choosing between YAML and Phenopacket input formats
How should users choose between YAML or Phenopacket as input?