Tutorial
This tutorial shows how to use LIRICAL to evaluate an exome.
Setup
Follow the instructions in Setting up LIRICAL to get LIRICAL executable, download the database files and the Exomiser database. Note the location of the Exomiser variant database file.
The data
We will run an exome analysis of a case reported in Cao et al., 2018. The authors describe a disease-associated mutation in the TGFBR2 gene in Patient 4.
We have simulated an exome VCF file by adding the disease associated variant to a VCF file derived from project.NIST.hc.snps.indels.NIST7035.vcf and we prepared a phenopacket to represent patient’s clinical data (see below). The files are available in the examples folder or in the distribution ZIP.
Creating a phenopacket
Creating the phenopacket is an optional step and the example phenopacket can be used for the purpose of this tutorial.
Here is an excerpt of the text that described patient 4 in the above cited article:
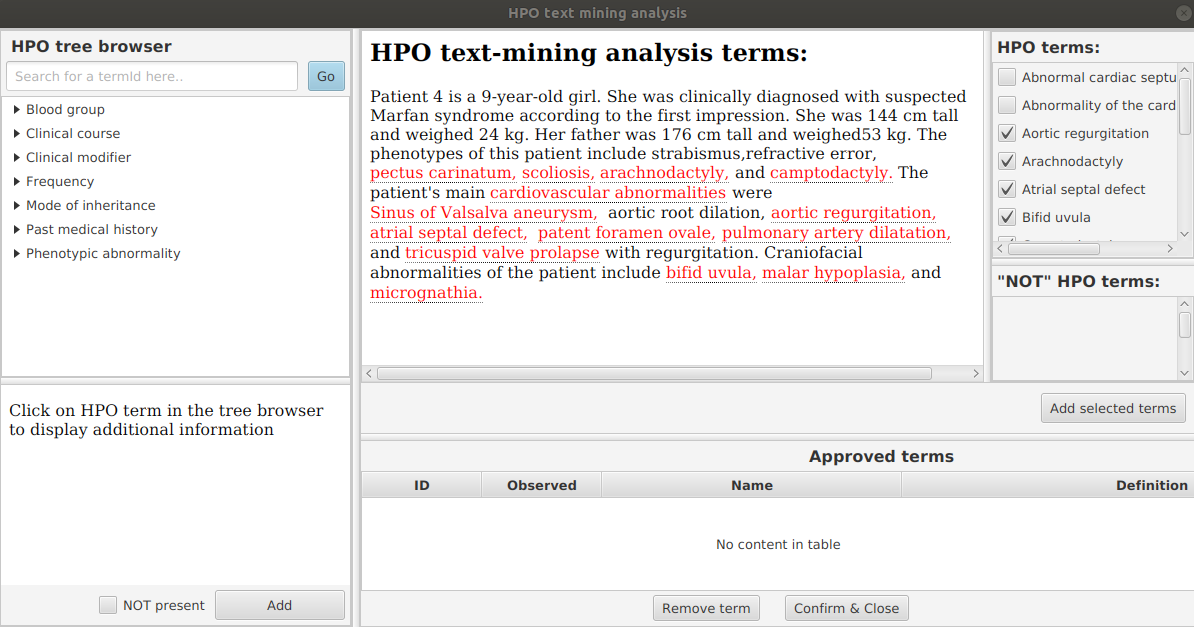
Patient 4 is a 9-year-old girl. She was clinically diagnosed with suspected
Marfan syndrome according to the first impression. She was 144 cm tall and
weighed 24 kg. Her father was 176 cm tall and weighed 53 kg. The phenotypes
of this patient include strabismus, refractive error, pectus carinatum, scoliosis,
arachnodactyly, and camptodactyly. The patient's main cardiovascular abnormalities
were Sinus of Valsalva aneurysm, aortic root dilation, aortic regurgitation,
atrial septal defect, patent foramen ovale, pulmonary artery dilatation, and
tricuspid valve prolapse with regurgitation. Craniofacial abnormalities of the
patient include bifid uvula, malar hypoplasia, and micrognathia.
Use the PhenopacketGenerator to create a Phenopacket.
To set up PhenopacketGenerator, you will first need to set the location of the hp.obo file from the Download page of the HPO website. Enter your Biocurator id by selecting “Set biocurator id” from the edit menu, and enter an arbitrary Phenopacket ID and proband ID. Use the dropdown menus to enter “9 years” for Age and “Female” for sex.
From the edit menu, select “Set path to hp.obo file”, then select the location of the hpo.obo on your computer. After a moment, the ontology will load and “Enter HPO terms” will be clickable. Load HPO terms for this case by clicking “Enter HPO term”. Paste the clinical description above into the text-mining window of PhenopacketGenerator, click “Analyze”, select HPO terms, click “Add selected terms”, then “Confirm and Close”.
Then, select the location of the VCF file that you saved in the previous step, and enter the Genome assembly (hg19).
You can now export the phenopacket. Use the filename LDS2.json
(or choose another name and adjust the following command accordingly).
Running LIRICAL
Run LIRICAL as follows:
lirical phenopacket \
--assembly hg19 \
--exomiser-hg19-dir /path/to/2406_hg19 \
--phenopacket LDS2.v2.json \
--vcf LDS2.vcf.gz \
--prefix LDS2
Note
We assume the LIRICAL alias was set as described in the Set up alias section.
Note
We assume that /path/to/2406_hg19 points to a folder with the 2406 Exomiser data release files.
Viewing the results
The above command will create a new file called LDS2.html (the -x | --prefix option controls the prefix of the output file).
Open this file in a web browser. The top of the page shows some information about the input files and a list of observed
and excluded HPO terms. The next section shows summarized representations of the top candidates.
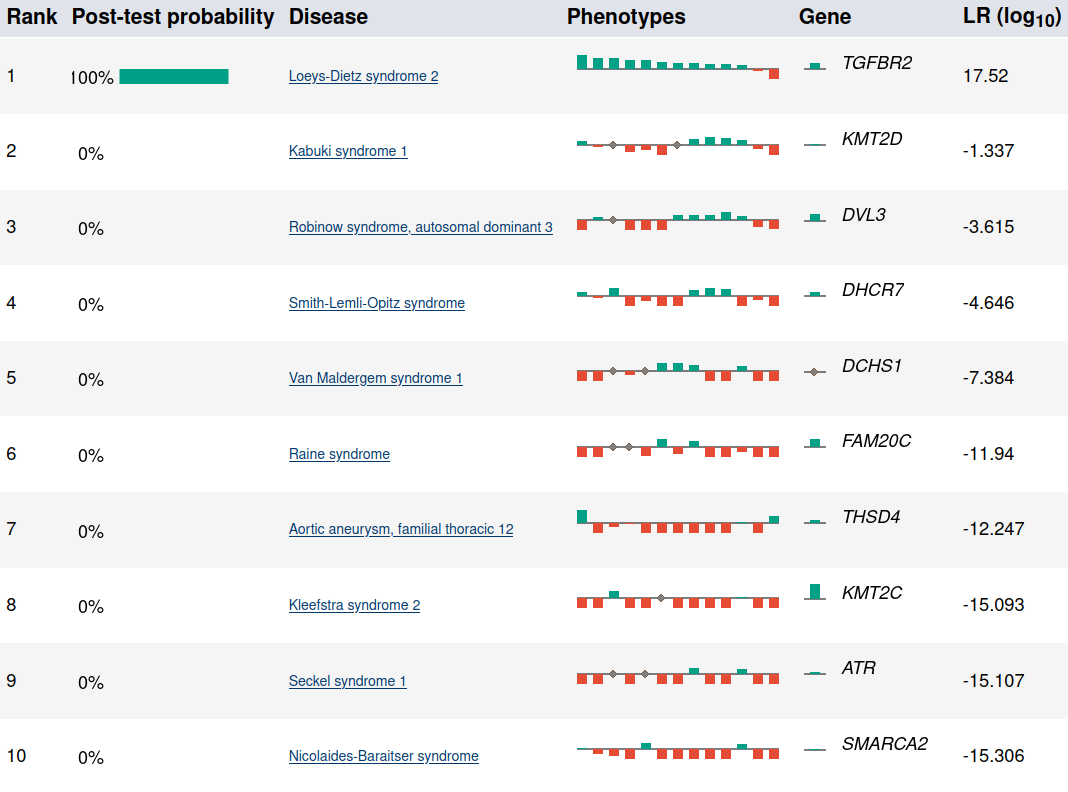
Summary view of the top candidates.
Each row in the summary shows the rank, post-test probability, and name/ID of the disease. The row includes a sparkline representation of the phenotypic profiles of each candidate, with green bars indicating positive contributions and red bars negative contributions to the diagnosis. The last bar represents the genotype likelihood ratio if LIRICAL was run with a VCF file. Hovering over the individual bars will show the name of the HPO term or gene, and all sparklines show the terms in the same order.
LIRICAL then presents a detailed analysis of each of the top candidates. The summary shows information about identified variants and the phenotypic profile. Hovering over the graphic shows information about the likelihood ratio and the type of the match.
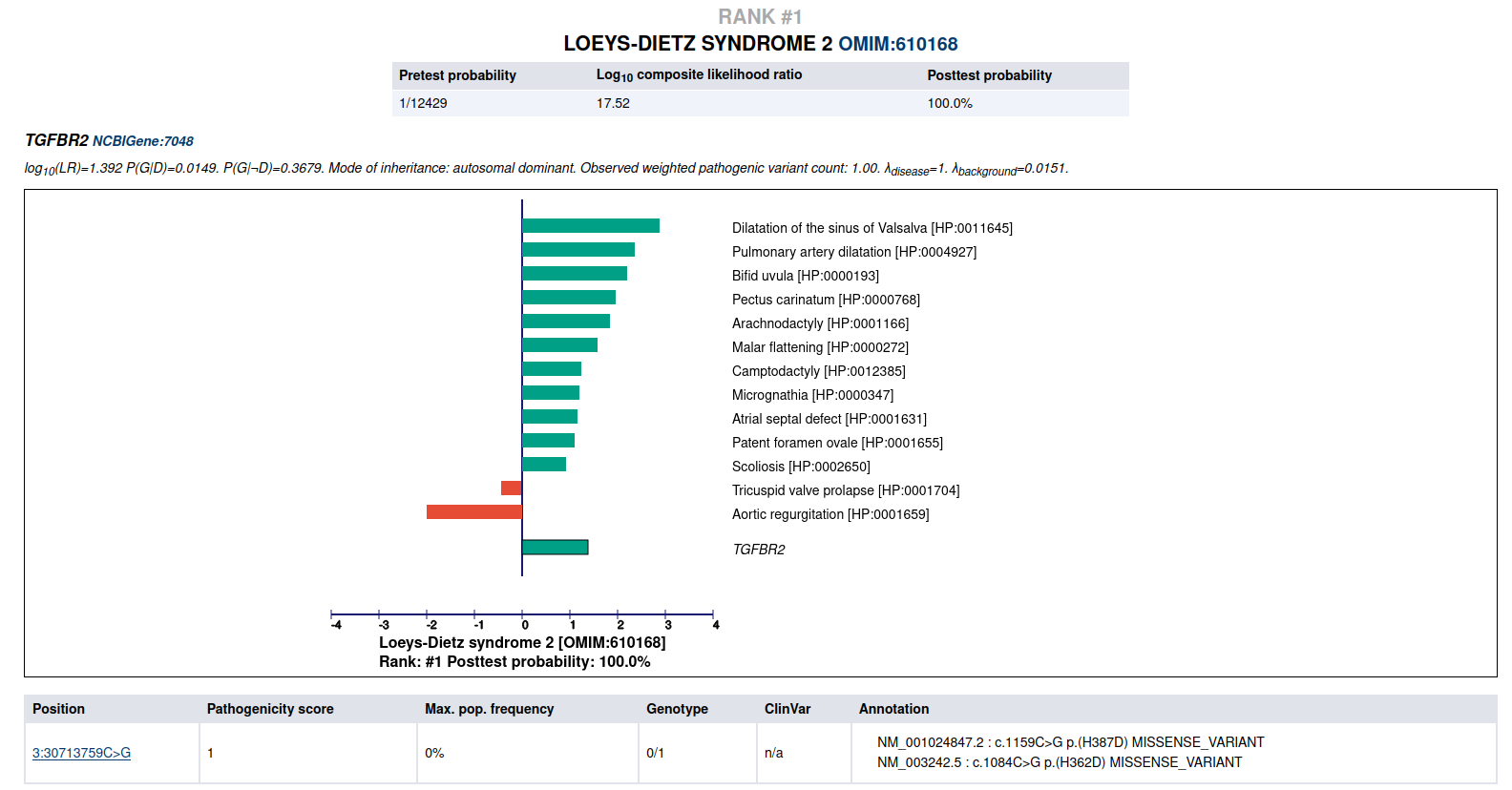
Detailed view of the top candidate Loeys-Dietz syndrome type 2.
The remaining part of the HTML output page contains information about the other top candidates and a list of all diseases analyzed. The bottom of the page includes explanations and documents the settings used for the analysis.