GUI – Data preparation
Overview
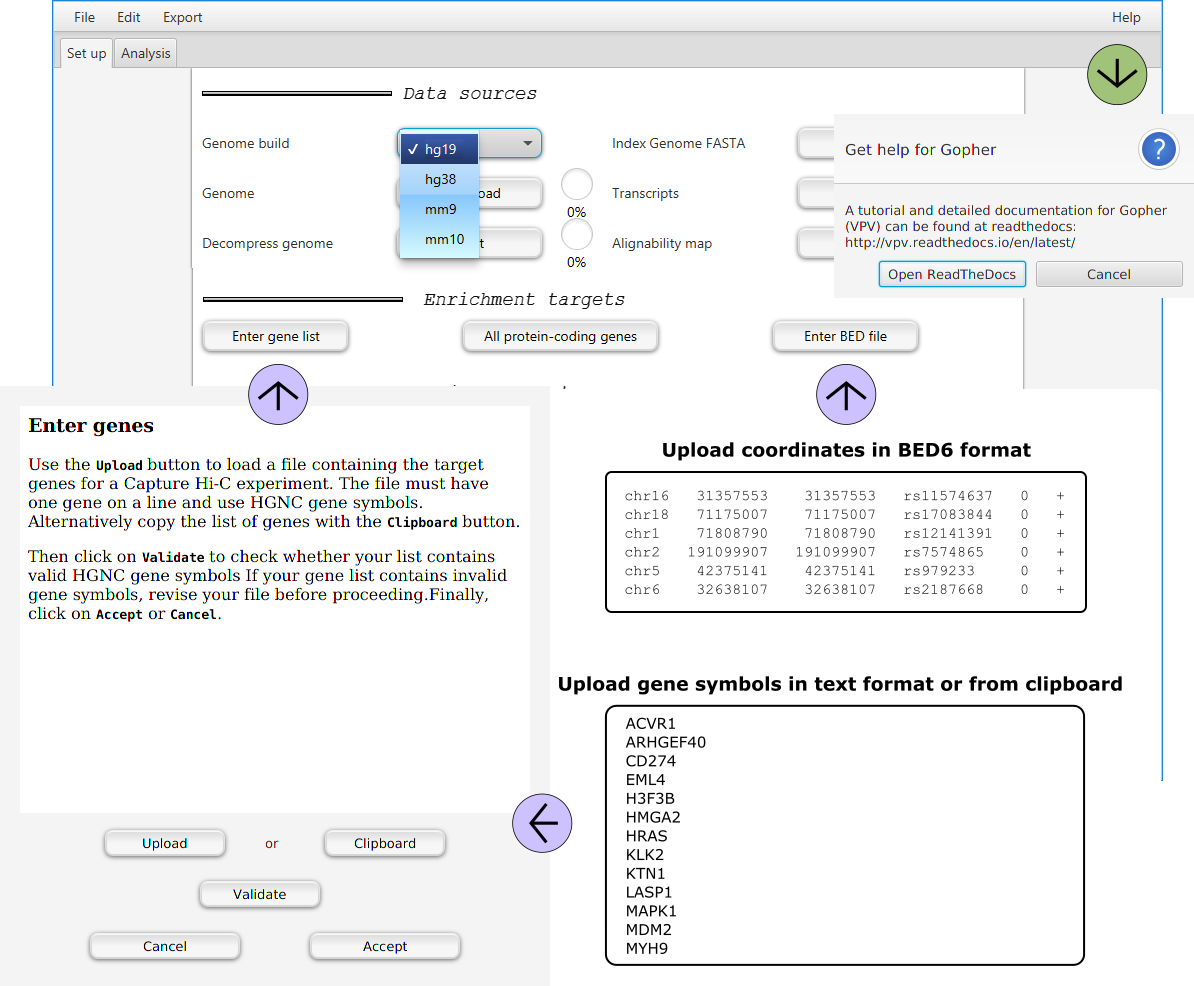
Sequence and annotation data for various genome builds for mouse and human can be downloaded and preprocessed via mouse clicks. Lists of target gene symbols can be uploaded from a text file or from the clipboard. Alternatively, a shortcut allows to select all protein-coding genes as capture targets. GOPHER’s functionality is not restricted to transcription start sites. Arbitrary genomic position can be uploaded in BED6 format, for instance GWAS hits.
Choosing a genome build
GOPHER currently supports hg19 (i.e., GRCh37), GRCh38, mm9 and mm10. Choose the appropriate genome build from the pulldown menu.
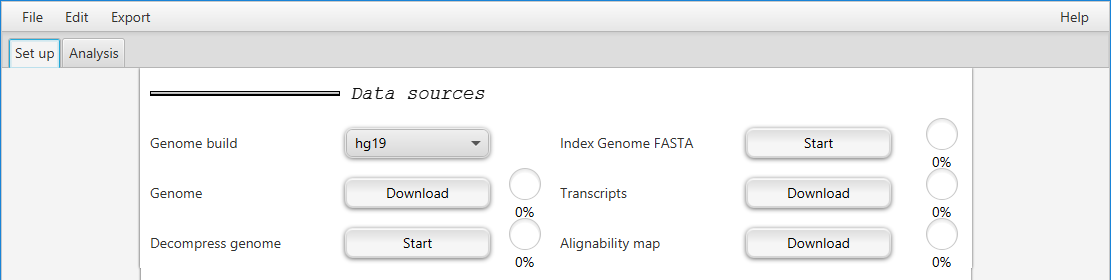
Click on the Download button and choose a directory for the genome.
GOPHER will download the corresponding genome file from the UCSC Genome Browser unless it finds the correspding files in the directory, in which case it will merely show the Done mark.
Note that the full download may take many minutes or longer depending on your network bandwidth.
The UCSC Genome files are provided as compressed (gzip) tar archives.
Clicking on the Start button after Decompress genome will decompress the files if necessary.
If GOPHER finds the unpacked files in the genome directory, it will show the Done mark immediately.
The genome FASTA files (one for each chromosome) need to be indexed.
GOPHER indexes the files to produce .fai index files that are equivalent to those produced by samtools.
If desired you can use samtools yourself.
If GOPHER finds the .fai files in the directory, it will show the Done mark immediately.
GOPHER additionally requires a transcript definition file.
It will automatically download the correct file from UCSC if the user clicks on the Download button after Transcripts.
It is recommended to store the file in the same directory as the genome file.
If GOPHER finds the file in the direcotry (refGene.txt.gz) it will show the Done mark immediately.
Finally, GOPHER needs to download the alignabilty map from UCSC.
For a given kmer size \(k\), the alignabilty map \(p \mapsto a_k(p)\) indicates how often the kmer starting at position \(p\) occurs within in the entire genome.
GOPHER will automatically download the it correct file from UCSC if the user clicks on the Download button after Alignabilty map.
Choosing enrichment targets
Following the steps described above, the user specifies the desired enrichment targets.
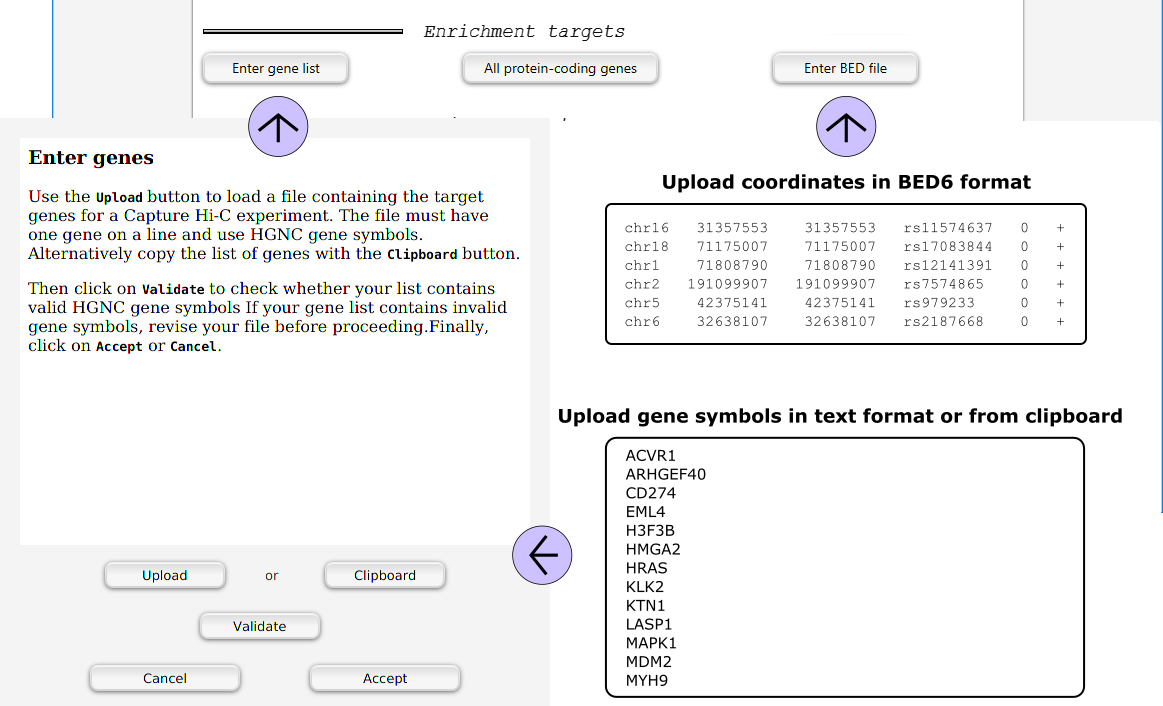
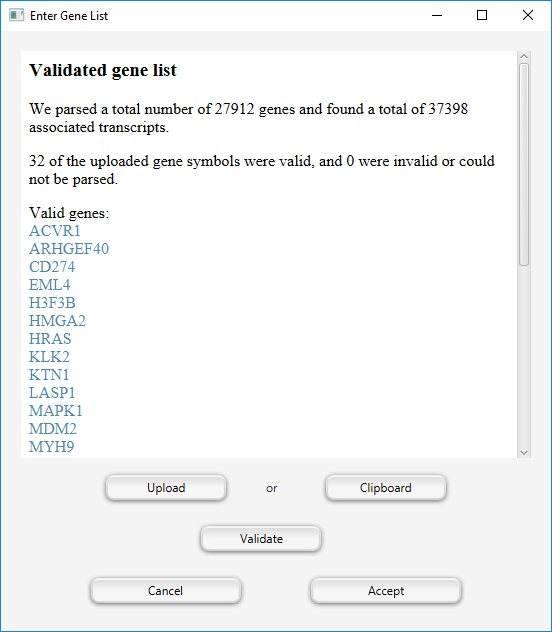
Clicking on the Enter gene list button will open a dialog to enter a gene list.
Currently, GOPHER expects a list of valid (HGNC) gene symbols.
For promoter CHC, gene symbols can be uploaded from a text file with the Target Gene List button.
Use the All protein-coding genes button to extract all protein-coding gene symbols from the transcript file.
Next, click the Validate button.
If gene symbols are used that do not occur in the downloaded annotation data, GOPHER will issue a warning and report a list of unmappable symbols that can be used to search for the current correct symbols.
Finally, click the Accept button.
Alternatively, the All protein-coding genes button allows promoters of all-protein coding genes to be selected as targets.
GOPHER also accepts a BED file with genomic positions.
For instance, the coordinates of GWAS hits can be uploaded in BED6 format.
Click the Enter BED file button and select the file.
GOPHER also has two sample gene lists for human and mouse genomes that can be used as enrichment targets.
To access the lists, click on Help > Example gene sets.
Select human or mouse and click Validate and Accept.