DEG_analysis_GSE100356
Last updated: 2022-10-02
Checks: 7 0
Knit directory: DO_Opioid/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200504) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7b78c2e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/Picture1.png
Untracked files:
Untracked: Rplot_rz.png
Untracked: TIMBR.test.prop.bm.MN.RET.RData
Untracked: TIMBR.test.random.RData
Untracked: TIMBR.test.rz.transformed_TVb_ml.RData
Untracked: analysis/DDO_morphine1_second_set_69k.stdout
Untracked: analysis/DO_Fentanyl.R
Untracked: analysis/DO_Fentanyl.err
Untracked: analysis/DO_Fentanyl.out
Untracked: analysis/DO_Fentanyl.sh
Untracked: analysis/DO_Fentanyl_69k.R
Untracked: analysis/DO_Fentanyl_69k.err
Untracked: analysis/DO_Fentanyl_69k.out
Untracked: analysis/DO_Fentanyl_69k.sh
Untracked: analysis/DO_Fentanyl_Cohort2_gemma.R
Untracked: analysis/DO_Fentanyl_Cohort2_mapping.R
Untracked: analysis/DO_Fentanyl_Cohort2_mapping.err
Untracked: analysis/DO_Fentanyl_Cohort2_mapping.out
Untracked: analysis/DO_Fentanyl_Cohort2_mapping.sh
Untracked: analysis/DO_Fentanyl_GCTA_herit.R
Untracked: analysis/DO_Fentanyl_alternate_metrics_69k.R
Untracked: analysis/DO_Fentanyl_alternate_metrics_69k.err
Untracked: analysis/DO_Fentanyl_alternate_metrics_69k.out
Untracked: analysis/DO_Fentanyl_alternate_metrics_69k.sh
Untracked: analysis/DO_Fentanyl_alternate_metrics_array.R
Untracked: analysis/DO_Fentanyl_alternate_metrics_array.err
Untracked: analysis/DO_Fentanyl_alternate_metrics_array.out
Untracked: analysis/DO_Fentanyl_alternate_metrics_array.sh
Untracked: analysis/DO_Fentanyl_array.R
Untracked: analysis/DO_Fentanyl_array.err
Untracked: analysis/DO_Fentanyl_array.out
Untracked: analysis/DO_Fentanyl_array.sh
Untracked: analysis/DO_Fentanyl_combining2Cohort_gemma.R
Untracked: analysis/DO_Fentanyl_combining2Cohort_mapping.R
Untracked: analysis/DO_Fentanyl_combining2Cohort_mapping.err
Untracked: analysis/DO_Fentanyl_combining2Cohort_mapping.out
Untracked: analysis/DO_Fentanyl_combining2Cohort_mapping.sh
Untracked: analysis/DO_Fentanyl_combining2Cohort_mapping_CoxPH.R
Untracked: analysis/DO_Fentanyl_finalreport_to_plink.sh
Untracked: analysis/DO_Fentanyl_gemma.R
Untracked: analysis/DO_Fentanyl_gemma.err
Untracked: analysis/DO_Fentanyl_gemma.out
Untracked: analysis/DO_Fentanyl_gemma.sh
Untracked: analysis/DO_morphine1.R
Untracked: analysis/DO_morphine1.Rout
Untracked: analysis/DO_morphine1.sh
Untracked: analysis/DO_morphine1.stderr
Untracked: analysis/DO_morphine1.stdout
Untracked: analysis/DO_morphine1_SNP.R
Untracked: analysis/DO_morphine1_SNP.Rout
Untracked: analysis/DO_morphine1_SNP.sh
Untracked: analysis/DO_morphine1_SNP.stderr
Untracked: analysis/DO_morphine1_SNP.stdout
Untracked: analysis/DO_morphine1_combined.R
Untracked: analysis/DO_morphine1_combined.Rout
Untracked: analysis/DO_morphine1_combined.sh
Untracked: analysis/DO_morphine1_combined.stderr
Untracked: analysis/DO_morphine1_combined.stdout
Untracked: analysis/DO_morphine1_combined_69k.R
Untracked: analysis/DO_morphine1_combined_69k.Rout
Untracked: analysis/DO_morphine1_combined_69k.sh
Untracked: analysis/DO_morphine1_combined_69k.stderr
Untracked: analysis/DO_morphine1_combined_69k.stdout
Untracked: analysis/DO_morphine1_combined_69k_m2.R
Untracked: analysis/DO_morphine1_combined_69k_m2.Rout
Untracked: analysis/DO_morphine1_combined_69k_m2.sh
Untracked: analysis/DO_morphine1_combined_69k_m2.stderr
Untracked: analysis/DO_morphine1_combined_69k_m2.stdout
Untracked: analysis/DO_morphine1_combined_weight_DOB.R
Untracked: analysis/DO_morphine1_combined_weight_DOB.Rout
Untracked: analysis/DO_morphine1_combined_weight_DOB.err
Untracked: analysis/DO_morphine1_combined_weight_DOB.out
Untracked: analysis/DO_morphine1_combined_weight_DOB.sh
Untracked: analysis/DO_morphine1_combined_weight_DOB.stderr
Untracked: analysis/DO_morphine1_combined_weight_DOB.stdout
Untracked: analysis/DO_morphine1_combined_weight_age.R
Untracked: analysis/DO_morphine1_combined_weight_age.err
Untracked: analysis/DO_morphine1_combined_weight_age.out
Untracked: analysis/DO_morphine1_combined_weight_age.sh
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT.R
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT.err
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT.out
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT.sh
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT_chr19.R
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT_chr19.err
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT_chr19.out
Untracked: analysis/DO_morphine1_combined_weight_age_GAMMT_chr19.sh
Untracked: analysis/DO_morphine1_cph.R
Untracked: analysis/DO_morphine1_cph.Rout
Untracked: analysis/DO_morphine1_cph.sh
Untracked: analysis/DO_morphine1_second_set.R
Untracked: analysis/DO_morphine1_second_set.Rout
Untracked: analysis/DO_morphine1_second_set.sh
Untracked: analysis/DO_morphine1_second_set.stderr
Untracked: analysis/DO_morphine1_second_set.stdout
Untracked: analysis/DO_morphine1_second_set_69k.R
Untracked: analysis/DO_morphine1_second_set_69k.Rout
Untracked: analysis/DO_morphine1_second_set_69k.sh
Untracked: analysis/DO_morphine1_second_set_69k.stderr
Untracked: analysis/DO_morphine1_second_set_SNP.R
Untracked: analysis/DO_morphine1_second_set_SNP.Rout
Untracked: analysis/DO_morphine1_second_set_SNP.sh
Untracked: analysis/DO_morphine1_second_set_SNP.stderr
Untracked: analysis/DO_morphine1_second_set_SNP.stdout
Untracked: analysis/DO_morphine1_second_set_weight_DOB.R
Untracked: analysis/DO_morphine1_second_set_weight_DOB.Rout
Untracked: analysis/DO_morphine1_second_set_weight_DOB.err
Untracked: analysis/DO_morphine1_second_set_weight_DOB.out
Untracked: analysis/DO_morphine1_second_set_weight_DOB.sh
Untracked: analysis/DO_morphine1_second_set_weight_DOB.stderr
Untracked: analysis/DO_morphine1_second_set_weight_DOB.stdout
Untracked: analysis/DO_morphine1_second_set_weight_age.R
Untracked: analysis/DO_morphine1_second_set_weight_age.Rout
Untracked: analysis/DO_morphine1_second_set_weight_age.err
Untracked: analysis/DO_morphine1_second_set_weight_age.out
Untracked: analysis/DO_morphine1_second_set_weight_age.sh
Untracked: analysis/DO_morphine1_second_set_weight_age.stderr
Untracked: analysis/DO_morphine1_second_set_weight_age.stdout
Untracked: analysis/DO_morphine1_weight_DOB.R
Untracked: analysis/DO_morphine1_weight_DOB.sh
Untracked: analysis/DO_morphine1_weight_age.R
Untracked: analysis/DO_morphine1_weight_age.sh
Untracked: analysis/DO_morphine_gemma.R
Untracked: analysis/DO_morphine_gemma.err
Untracked: analysis/DO_morphine_gemma.out
Untracked: analysis/DO_morphine_gemma.sh
Untracked: analysis/DO_morphine_gemma_firstmin.R
Untracked: analysis/DO_morphine_gemma_firstmin.err
Untracked: analysis/DO_morphine_gemma_firstmin.out
Untracked: analysis/DO_morphine_gemma_firstmin.sh
Untracked: analysis/DO_morphine_gemma_withpermu.R
Untracked: analysis/DO_morphine_gemma_withpermu.err
Untracked: analysis/DO_morphine_gemma_withpermu.out
Untracked: analysis/DO_morphine_gemma_withpermu.sh
Untracked: analysis/DO_morphine_gemma_withpermu_firstbatch_min.depression.R
Untracked: analysis/DO_morphine_gemma_withpermu_firstbatch_min.depression.err
Untracked: analysis/DO_morphine_gemma_withpermu_firstbatch_min.depression.out
Untracked: analysis/DO_morphine_gemma_withpermu_firstbatch_min.depression.sh
Untracked: analysis/Plot_DO_morphine1_SNP.R
Untracked: analysis/Plot_DO_morphine1_SNP.Rout
Untracked: analysis/Plot_DO_morphine1_SNP.sh
Untracked: analysis/Plot_DO_morphine1_SNP.stderr
Untracked: analysis/Plot_DO_morphine1_SNP.stdout
Untracked: analysis/Plot_DO_morphine1_second_set_SNP.R
Untracked: analysis/Plot_DO_morphine1_second_set_SNP.Rout
Untracked: analysis/Plot_DO_morphine1_second_set_SNP.sh
Untracked: analysis/Plot_DO_morphine1_second_set_SNP.stderr
Untracked: analysis/Plot_DO_morphine1_second_set_SNP.stdout
Untracked: analysis/download_GSE100356_sra.sh
Untracked: analysis/fentanyl_2cohorts_coxph.R
Untracked: analysis/fentanyl_2cohorts_coxph.err
Untracked: analysis/fentanyl_2cohorts_coxph.out
Untracked: analysis/fentanyl_2cohorts_coxph.sh
Untracked: analysis/fentanyl_scanone.cph.R
Untracked: analysis/fentanyl_scanone.cph.err
Untracked: analysis/fentanyl_scanone.cph.out
Untracked: analysis/fentanyl_scanone.cph.sh
Untracked: analysis/geo_rnaseq.R
Untracked: analysis/heritability_first_second_batch.R
Untracked: analysis/nf-rnaseq-b6.R
Untracked: analysis/plot_fentanyl_2cohorts_coxph.R
Untracked: analysis/scripts/
Untracked: analysis/tibmr.R
Untracked: analysis/timbr_demo.R
Untracked: analysis/workflow_proc.R
Untracked: analysis/workflow_proc.sh
Untracked: analysis/workflow_proc.stderr
Untracked: analysis/workflow_proc.stdout
Untracked: analysis/x.R
Untracked: code/cfw/
Untracked: code/gemma_plot.R
Untracked: code/process.sanger.snp.R
Untracked: code/reconst_utils.R
Untracked: data/69k_grid_pgmap.RData
Untracked: data/Composite Post Kevins Program Group 2 Fentanyl Prepped for Hao.xlsx
Untracked: data/DO_WBP_Data_JAB_to_map.xlsx
Untracked: data/Fentanyl_alternate_metrics.xlsx
Untracked: data/FinalReport/
Untracked: data/GM/
Untracked: data/GM_covar.csv
Untracked: data/GM_covar_07092018_morphine.csv
Untracked: data/Jackson_Lab_Bubier_MURGIGV01/
Untracked: data/MPD_Upload_October.csv
Untracked: data/MPD_Upload_October_updated_sex.csv
Untracked: data/Master Fentanyl DO Study Sheet.xlsx
Untracked: data/MasterMorphine Second Set DO w DOB2.xlsx
Untracked: data/MasterMorphine Second Set DO.xlsx
Untracked: data/Morphine CC DO mice Updated with Published inbred strains.csv
Untracked: data/Morphine_CC_DO_mice_Updated_with_Published_inbred_strains.csv
Untracked: data/cc_variants.sqlite
Untracked: data/combined/
Untracked: data/fentanyl/
Untracked: data/fentanyl2/
Untracked: data/fentanyl_1_2/
Untracked: data/fentanyl_2cohorts_coxph_data.Rdata
Untracked: data/first/
Untracked: data/founder_geno.csv
Untracked: data/genetic_map.csv
Untracked: data/gm.json
Untracked: data/gwas.sh
Untracked: data/marker_grid_0.02cM_plus.txt
Untracked: data/mouse_genes_mgi.sqlite
Untracked: data/pheno.csv
Untracked: data/pheno_qtl2.csv
Untracked: data/pheno_qtl2_07092018_morphine.csv
Untracked: data/pheno_qtl2_w_dob.csv
Untracked: data/physical_map.csv
Untracked: data/rnaseq/
Untracked: data/sample_geno.csv
Untracked: data/second/
Untracked: figure/
Untracked: glimma-plots/
Untracked: output/DO_Fentanyl_Cohort2_MinDepressionRR_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_MinDepressionRR_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_RRRecoveryRateHrSLOPE_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_RRRecoveryRateHrSLOPE_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_StartofRecoveryHr_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_StartofRecoveryHr_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_Statusbin_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_Statusbin_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoDead(Hr)_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoDeadHr_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoDeadHr_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoProjectedRecoveryHr_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoProjectedRecoveryHr_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoSteadyRRDepression(Hr)_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoSteadyRRDepressionHr_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoSteadyRRDepressionHr_coefplot_blup.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoThresholdRecoveryHr_coefplot.pdf
Untracked: output/DO_Fentanyl_Cohort2_TimetoThresholdRecoveryHr_coefplot_blup.pdf
Untracked: output/DO_morphine_Min.depression.png
Untracked: output/DO_morphine_Min.depression22222_violin_chr5.pdf
Untracked: output/DO_morphine_Min.depression_coefplot.pdf
Untracked: output/DO_morphine_Min.depression_coefplot_blup.pdf
Untracked: output/DO_morphine_Min.depression_coefplot_blup_chr5.png
Untracked: output/DO_morphine_Min.depression_coefplot_blup_chrX.png
Untracked: output/DO_morphine_Min.depression_coefplot_chr5.png
Untracked: output/DO_morphine_Min.depression_coefplot_chrX.png
Untracked: output/DO_morphine_Min.depression_peak_genes_chr5.png
Untracked: output/DO_morphine_Min.depression_violin_chr5.png
Untracked: output/DO_morphine_Recovery.Time.png
Untracked: output/DO_morphine_Recovery.Time_coefplot.pdf
Untracked: output/DO_morphine_Recovery.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_Recovery.Time_coefplot_blup_chr11.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_blup_chr4.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_blup_chr7.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_blup_chr9.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_chr11.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_chr4.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_chr7.png
Untracked: output/DO_morphine_Recovery.Time_coefplot_chr9.png
Untracked: output/DO_morphine_Status_bin.png
Untracked: output/DO_morphine_Status_bin_coefplot.pdf
Untracked: output/DO_morphine_Status_bin_coefplot_blup.pdf
Untracked: output/DO_morphine_Survival.Time.png
Untracked: output/DO_morphine_Survival.Time_coefplot.pdf
Untracked: output/DO_morphine_Survival.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_Survival.Time_coefplot_blup_chr17.png
Untracked: output/DO_morphine_Survival.Time_coefplot_blup_chr8.png
Untracked: output/DO_morphine_Survival.Time_coefplot_chr17.png
Untracked: output/DO_morphine_Survival.Time_coefplot_chr8.png
Untracked: output/DO_morphine_combine_batch_peak_violin.pdf
Untracked: output/DO_morphine_combined_69k_m2_Min.depression.png
Untracked: output/DO_morphine_combined_69k_m2_Min.depression_coefplot.pdf
Untracked: output/DO_morphine_combined_69k_m2_Min.depression_coefplot_blup.pdf
Untracked: output/DO_morphine_combined_69k_m2_Recovery.Time.png
Untracked: output/DO_morphine_combined_69k_m2_Recovery.Time_coefplot.pdf
Untracked: output/DO_morphine_combined_69k_m2_Recovery.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_combined_69k_m2_Status_bin.png
Untracked: output/DO_morphine_combined_69k_m2_Status_bin_coefplot.pdf
Untracked: output/DO_morphine_combined_69k_m2_Status_bin_coefplot_blup.pdf
Untracked: output/DO_morphine_combined_69k_m2_Survival.Time.png
Untracked: output/DO_morphine_combined_69k_m2_Survival.Time_coefplot.pdf
Untracked: output/DO_morphine_combined_69k_m2_Survival.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_coxph_24hrs_kinship_QTL.png
Untracked: output/DO_morphine_cphout.RData
Untracked: output/DO_morphine_first_batch_peak_in_second_batch_violin.pdf
Untracked: output/DO_morphine_first_batch_peak_in_second_batch_violin_sidebyside.pdf
Untracked: output/DO_morphine_first_batch_peak_violin.pdf
Untracked: output/DO_morphine_operm.cph.RData
Untracked: output/DO_morphine_second_batch_on_first_batch_peak_violin.pdf
Untracked: output/DO_morphine_second_batch_peak_ch6surv_on_first_batchviolin.pdf
Untracked: output/DO_morphine_second_batch_peak_ch6surv_on_first_batchviolin2.pdf
Untracked: output/DO_morphine_second_batch_peak_in_first_batch_violin.pdf
Untracked: output/DO_morphine_second_batch_peak_in_first_batch_violin_sidebyside.pdf
Untracked: output/DO_morphine_second_batch_peak_violin.pdf
Untracked: output/DO_morphine_secondbatch_69k_Min.depression.png
Untracked: output/DO_morphine_secondbatch_69k_Min.depression_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_69k_Min.depression_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_69k_Recovery.Time.png
Untracked: output/DO_morphine_secondbatch_69k_Recovery.Time_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_69k_Recovery.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_69k_Status_bin.png
Untracked: output/DO_morphine_secondbatch_69k_Status_bin_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_69k_Status_bin_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_69k_Survival.Time.png
Untracked: output/DO_morphine_secondbatch_69k_Survival.Time_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_69k_Survival.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_Min.depression.png
Untracked: output/DO_morphine_secondbatch_Min.depression_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_Min.depression_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_Recovery.Time.png
Untracked: output/DO_morphine_secondbatch_Recovery.Time_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_Recovery.Time_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_Status_bin.png
Untracked: output/DO_morphine_secondbatch_Status_bin_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_Status_bin_coefplot_blup.pdf
Untracked: output/DO_morphine_secondbatch_Survival.Time.png
Untracked: output/DO_morphine_secondbatch_Survival.Time_coefplot.pdf
Untracked: output/DO_morphine_secondbatch_Survival.Time_coefplot_blup.pdf
Untracked: output/Fentanyl/
Untracked: output/TIMBR.test.RData
Untracked: output/apr_69kchr_combined.RData
Untracked: output/apr_69kchr_k_loco_combined.rds
Untracked: output/apr_69kchr_second_set.RData
Untracked: output/combine_batch_variation.RData
Untracked: output/combined_gm.RData
Untracked: output/combined_gm.k_loco.rds
Untracked: output/combined_gm.k_overall.rds
Untracked: output/combined_gm.probs_8state.rds
Untracked: output/coxph/
Untracked: output/do.morphine.RData
Untracked: output/do.morphine.k_loco.rds
Untracked: output/do.morphine.probs_36state.rds
Untracked: output/do.morphine.probs_8state.rds
Untracked: output/do_Fentanyl_combine2cohort_MeanDepressionBR_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_MeanDepressionBR_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_MinDepressionBR_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_MinDepressionBR_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_MinDepressionRR_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_MinDepressionRR_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_RRRecoveryRateHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_RRRecoveryRateHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_StartofRecoveryHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_StartofRecoveryHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_Statusbin_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_Statusbin_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_SteadyStateDepressionDurationHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_SteadyStateDepressionDurationHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_SurvivalTime_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_SurvivalTime_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoDeadHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoDeadHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoMostlyDeadHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoMostlyDeadHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoProjectedRecoveryHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoProjectedRecoveryHr_coefplot_blup.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoRecoveryHr_coefplot.pdf
Untracked: output/do_Fentanyl_combine2cohort_TimetoRecoveryHr_coefplot_blup.pdf
Untracked: output/first_batch_variation.RData
Untracked: output/first_second_survival_peak_chr.xlsx
Untracked: output/hsq_1_first_batch_herit_qtl2.RData
Untracked: output/hsq_2_second_batch_herit_qtl2.RData
Untracked: output/old_temp/
Untracked: output/pr_69kchr_combined.RData
Untracked: output/pr_69kchr_second_set.RData
Untracked: output/qtl.morphine.69k.out.combined.RData
Untracked: output/qtl.morphine.69k.out.combined_m2.RData
Untracked: output/qtl.morphine.69k.out.second_set.RData
Untracked: output/qtl.morphine.operm.RData
Untracked: output/qtl.morphine.out.RData
Untracked: output/qtl.morphine.out.combined_gm.RData
Untracked: output/qtl.morphine.out.combined_weight_DOB.RData
Untracked: output/qtl.morphine.out.combined_weight_age.RData
Untracked: output/qtl.morphine.out.second_set.RData
Untracked: output/qtl.morphine.out.second_set.weight_DOB.RData
Untracked: output/qtl.morphine.out.second_set.weight_age.RData
Untracked: output/qtl.morphine.out.weight_DOB.RData
Untracked: output/qtl.morphine.out.weight_age.RData
Untracked: output/qtl.morphine1.snpout.RData
Untracked: output/qtl.morphine2.snpout.RData
Untracked: output/second_batch_pheno.csv
Untracked: output/second_batch_variation.RData
Untracked: output/second_set_apr_69kchr_k_loco.rds
Untracked: output/second_set_gm.RData
Untracked: output/second_set_gm.k_loco.rds
Untracked: output/second_set_gm.probs_36state.rds
Untracked: output/second_set_gm.probs_8state.rds
Untracked: output/topSNP_chr5_mindepression.csv
Untracked: output/zoompeak_Min.depression_9.pdf
Untracked: output/zoompeak_Recovery.Time_16.pdf
Untracked: output/zoompeak_Status_bin_11.pdf
Untracked: output/zoompeak_Survival.Time_1.pdf
Untracked: output/zoompeak_fentanyl_Survival.Time_2.pdf
Untracked: sra-tools_v2.10.7.sif
Unstaged changes:
Modified: _workflowr.yml
Modified: analysis/Plot_DO_Fentanyl_combining2Cohort_mapping.Rmd
Modified: analysis/marker_violin.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/DEG_analysis_GSE100356.Rmd) and HTML (docs/DEG_analysis_GSE100356.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7b78c2e | xhyuo | 2022-10-02 | add download csv button DEG_analysis_GSE100356 |
| html | bc47d57 | xhyuo | 2022-09-29 | Build site. |
| Rmd | a5214f4 | xhyuo | 2022-09-29 | DEG_analysis_GSE100356.Rmd |
Compare RNA-seq data between GSE100356(B6) and NOD_Bot
Library
library(stringr)
library(tidyverse)Warning: replacing previous import 'lifecycle::last_warnings' by
'rlang::last_warnings' when loading 'hms'Warning: replacing previous import 'ellipsis::check_dots_unnamed' by
'rlang::check_dots_unnamed' when loading 'hms'Warning: replacing previous import 'ellipsis::check_dots_used' by
'rlang::check_dots_used' when loading 'hms'Warning: replacing previous import 'ellipsis::check_dots_empty' by
'rlang::check_dots_empty' when loading 'hms'Warning: package 'purrr' was built under R version 4.0.5library(edgeR)
library(limma)
library(Glimma)
library(gplots)
library(org.Mm.eg.db)
library(RColorBrewer)
library(DESeq2)
library(pheatmap)
library(ggrepel)
library(DT)
library(enrichR)
library(cowplot)
library(ggplotify)
library(sva)
set.seed(123)Read the genes results from nf-rnaseq.
Details in “analysis/download_GSE100356_sra.sh” /home/heh/cs-nf-pipelines/run.sh /home/heh/cs-nf-pipelines/run_jason.sh
#list all the file ending with *.genes.results
all.genes.results <- list.files(path = "/fastscratch/heh",
pattern = "*.genes.results",
full.names = TRUE,
all.files = TRUE,
recursive = TRUE)
all.genes.results <- all.genes.results[1:29]
#copy to folder /projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq
command.cp <- paste(paste0("cp ", all.genes.results, " /projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq"),
collapse = ";")
system(command.cp)
#all the file in the folder
all.genes.results <- list.files(path = "/projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq",
pattern = "*.genes.results",
full.names = FALSE,
all.files = TRUE,
recursive = TRUE)
#get the sample id
sampleid <- sub("_GT20-.*", "", all.genes.results)
sampleid <- sub("_pass.genes.results", "", sampleid)
#replace SRR id to GSM id
sampleid[24:29] <- paste0("GSM267944", 0:5)
#data.frame
df <- data.frame(file = all.genes.results, id = sampleid)
#merge expected_count column
command.merge <- paste0("cd /projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq; bash -c 'paste ", paste(paste0("<(cut -f 5 ", all.genes.results,")"), collapse = " "), " > /projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq/merged_expected_count'")
system(command.merge)
#read into R
merged_expected_count <- read.table("/projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq/merged_expected_count",header=T,sep="\t")
colnames(merged_expected_count) <- sampleid
expgene <- read.table(file = paste0("/projects/csna/rnaseq/bubier_inbred_rnaseq/nf-rnaseq/",
as.character(all.genes.results[[1]])),header=T,sep="\t")
rownames(merged_expected_count) <- expgene[,1]
write.csv(merged_expected_count,file="data/rnaseq/merged_expected_count.csv",quote=F,row.names=T)
save(merged_expected_count, file="data/rnaseq/merged_expected_count.RData")Count matrix and design matrix
load("data/rnaseq/merged_expected_count.RData")
design.matrix <- read.csv(file = "data/rnaseq/bubier_inbred_rnaseq_design_matrix.csv", header = TRUE, stringsAsFactors = F)
design.matrix <- design.matrix %>%
dplyr::mutate(id = sub("_GT20-.*", "", R1), .after = Mouse) %>%
dplyr::select(Mouse, id, Strain, Tissue) %>%
bind_rows(data.frame(Mouse = paste0("B",1:6),
id = paste0("GSM267944", 0:5),
Strain = "B6",
Tissue = c(rep("neurons", 6)))) %>%
dplyr::mutate(Batch = c(rep("batch1", 23), rep("batch2", 6))) %>%
unite("Strain_Tissue", Strain:Tissue, remove = FALSE) %>%
dplyr::mutate(across(Strain:Strain_Tissue, as.factor))
rownames(design.matrix) <- paste(design.matrix$Mouse, design.matrix$Strain, design.matrix$Tissue, sep = "_")
#order
all.equal(design.matrix$id, colnames(merged_expected_count))[1] TRUEcolnames(merged_expected_count) = rownames(design.matrix)
#To now construct the DESeqDataSet object from the matrix of counts and the sample information table, we use:
#floor countdata
countdata <- merged_expected_count
countdata <- floor(countdata)
#Removing genes that are lowly expressed as 0
countdata <- countdata[rowSums(countdata) != 0,]
#perform the batch correction
adjusted_countdata <- ComBat_seq(counts = as.matrix(countdata), batch = design.matrix$Batch)Found 2 batches
Using null model in ComBat-seq.
Adjusting for 0 covariate(s) or covariate level(s)
Estimating dispersions
Fitting the GLM model
Shrinkage off - using GLM estimates for parameters
Adjusting the data#DESeqDataSet
ddsMat <- DESeqDataSetFromMatrix(countData = adjusted_countdata,
colData = design.matrix,
design = ~Strain_Tissue)converting counts to integer modeQC process
#Pre-filtering the dataset
#perform a minimal pre-filtering to keep only rows that have at least 10 reads total.
keep <- rowSums(counts(ddsMat)) >= 10
ddsMat <- ddsMat[keep,]
ddsMatclass: DESeqDataSet
dim: 31329 29
metadata(1): version
assays(1): counts
rownames(31329): ENSMUSG00000000001_Gnai3 ENSMUSG00000000028_Cdc45 ...
ENSMUSG00000118653_AC159819.1 ENSMUSG00000118655_AC156032.1
rowData names(0):
colnames(29): M1_WSB_EiJ_Bot M1_NOD_ShiLtJ_NTS ... B5_B6_neurons
B6_B6_neurons
colData names(6): Mouse id ... Tissue Batch# DESeq2 creates a matrix when you use the counts() function
## First convert normalized_counts to a data frame and transfer the row names to a new column called "gene"
# this gives log2(n + 1)
ntd <- normTransform(ddsMat)
normalized_counts <- assay(ntd) %>%
data.frame() %>%
rownames_to_column(var="gene") %>%
as_tibble()
#The variance stabilizing transformation and the rlog
#The rlog tends to work well on small datasets (n < 30), potentially outperforming the VST when there is a wide range of sequencing depth across samples (an order of magnitude difference).
rld <- rlog(ddsMat, blind = FALSE)
head(assay(rld), 3) M1_WSB_EiJ_Bot M1_NOD_ShiLtJ_NTS M1_WSB_EiJ_NTS
ENSMUSG00000000001_Gnai3 9.7196966 10.301944 9.655086
ENSMUSG00000000028_Cdc45 3.0605738 5.540395 4.780168
ENSMUSG00000000031_H19 0.6916934 5.067076 4.390860
M2_NOD_ShiLtJ_Bot M2_WSB_EiJ_Bot M2_NOD_ShiLtJ_NTS
ENSMUSG00000000001_Gnai3 9.845226 9.449549 10.352407
ENSMUSG00000000028_Cdc45 1.560338 5.609662 5.492868
ENSMUSG00000000031_H19 1.230868 4.770416 5.241781
M2_WSB_EiJ_NTS M3_NOD_ShiLtJ_Bot M3_WSB_EiJ_Bot
ENSMUSG00000000001_Gnai3 9.930522 10.528802 9.313502
ENSMUSG00000000028_Cdc45 1.100719 4.975581 3.554421
ENSMUSG00000000031_H19 4.415029 3.831132 2.810500
M3_NOD_ShiLtJ_NTS M3_WSB_EiJ_NTS M4_WSB_EiJ_Bot
ENSMUSG00000000001_Gnai3 10.464754 9.767729 9.932096
ENSMUSG00000000028_Cdc45 6.028096 2.702331 3.765424
ENSMUSG00000000031_H19 6.093495 4.419805 4.683346
M4_NOD_ShiLtJ_Bot M4_WSB_EiJ_NTS M4_NOD_ShiLtJ_NTS
ENSMUSG00000000001_Gnai3 9.773286 10.385077 10.065988
ENSMUSG00000000028_Cdc45 5.643407 4.481560 5.642105
ENSMUSG00000000031_H19 4.951669 5.880698 5.107677
M5_WSB_EiJ_Bot M5_NOD_ShiLtJ_Bot M5_WSB_EiJ_NTS
ENSMUSG00000000001_Gnai3 9.972134 9.721972 9.597306
ENSMUSG00000000028_Cdc45 5.220241 2.592403 6.463934
ENSMUSG00000000031_H19 4.749526 3.360242 4.411733
M5_NOD_ShiLtJ_NTS M6_NOD_ShiLtJ_Bot M6_WSB_EiJ_Bot
ENSMUSG00000000001_Gnai3 9.950188 10.194515 9.793704
ENSMUSG00000000028_Cdc45 5.825242 5.003696 2.516536
ENSMUSG00000000031_H19 3.152996 3.374983 3.377736
M6_WSB_EiJ_NTS M6_NOD_ShiLtJ_NTS B1_B6_neurons
ENSMUSG00000000001_Gnai3 10.007504 9.886813 10.289235
ENSMUSG00000000028_Cdc45 5.351226 6.212049 2.276503
ENSMUSG00000000031_H19 4.455702 4.510894 3.822414
B2_B6_neurons B3_B6_neurons B4_B6_neurons
ENSMUSG00000000001_Gnai3 9.729727 10.059064 9.614295
ENSMUSG00000000028_Cdc45 5.183244 6.263450 5.739164
ENSMUSG00000000031_H19 3.184101 5.468361 5.376940
B5_B6_neurons B6_B6_neurons
ENSMUSG00000000001_Gnai3 10.215792 9.540164
ENSMUSG00000000028_Cdc45 5.422627 2.157702
ENSMUSG00000000031_H19 5.039048 4.054536#sample distance
sampleDists <- dist(t(assay(rld)))
sampleDistMatrix <- as.matrix( sampleDists )
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
#annotation
df <- as.data.frame(colData(ddsMat)[,c("Strain_Tissue")])
colnames(df) = "Strain_Tissue"
rownames(df) = rownames(sampleDistMatrix)
#heatmap on sample distance
pheatmap(sampleDistMatrix,
clustering_distance_rows = sampleDists,
clustering_distance_cols = sampleDists,
col = colors,
annotation_col = df,
border_color = NA)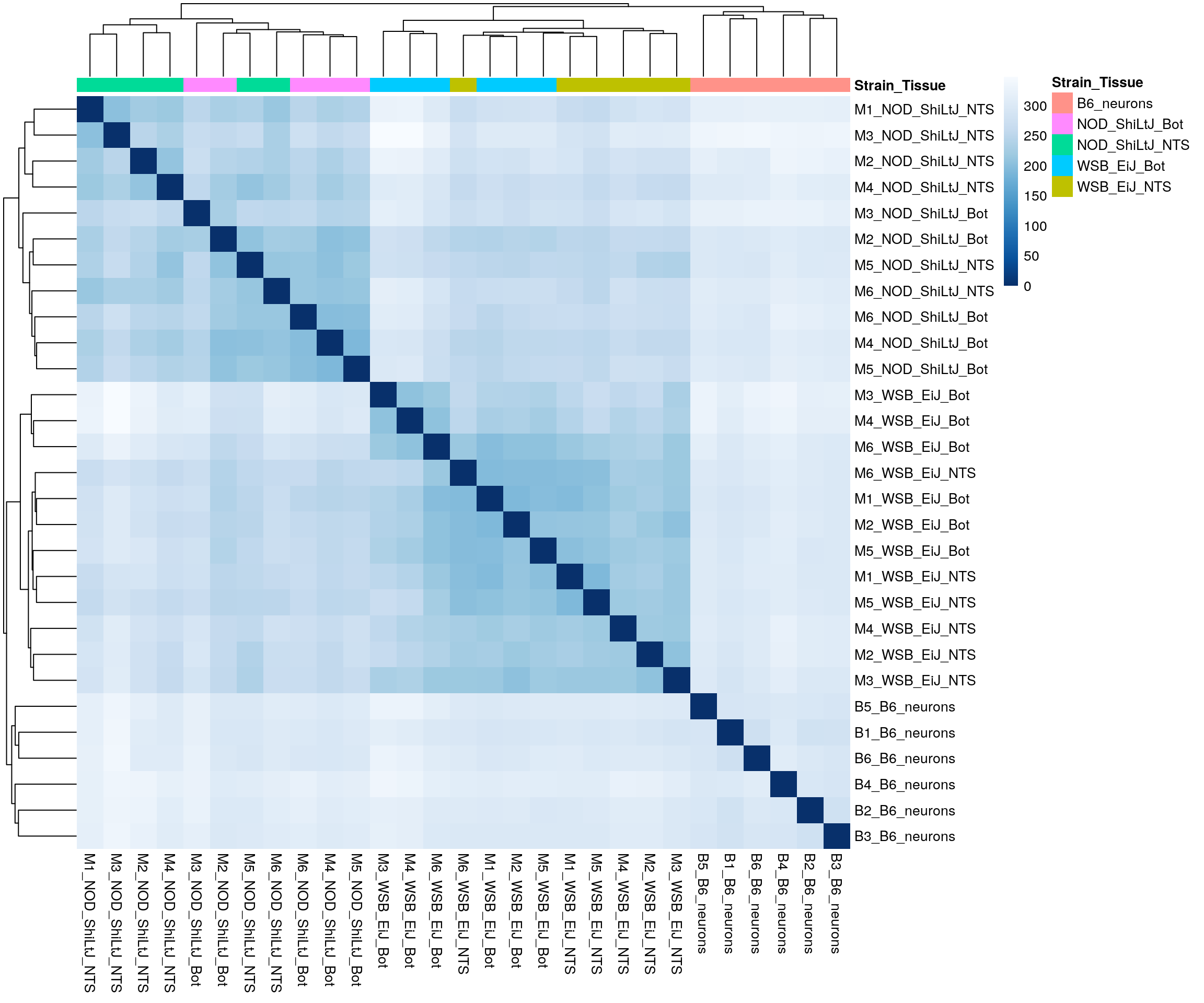
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
#heatmap on correlation matrix
rld_cor <- cor(assay(rld))
pheatmap(rld_cor,
annotation_col = df,
clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation",
border_color = NA)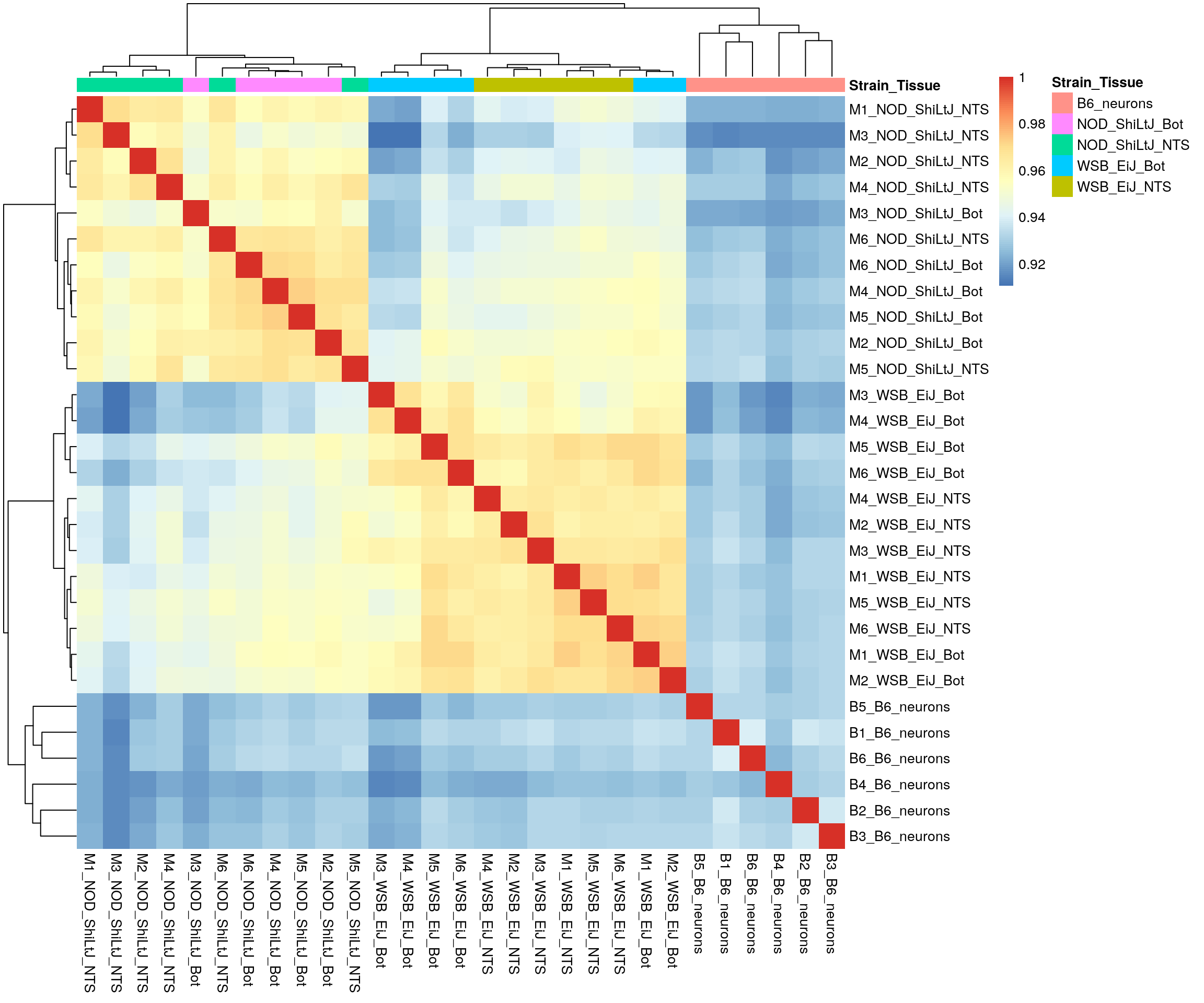
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
#PCA plot
#Another way to visualize sample-to-sample distances is a principal components analysis (PCA).
pca.plot <- plotPCA(rld, intgroup = c("Strain_Tissue"), returnData = FALSE)
pca.plot$data$name = ""
pca.plot + geom_text(aes(label=name))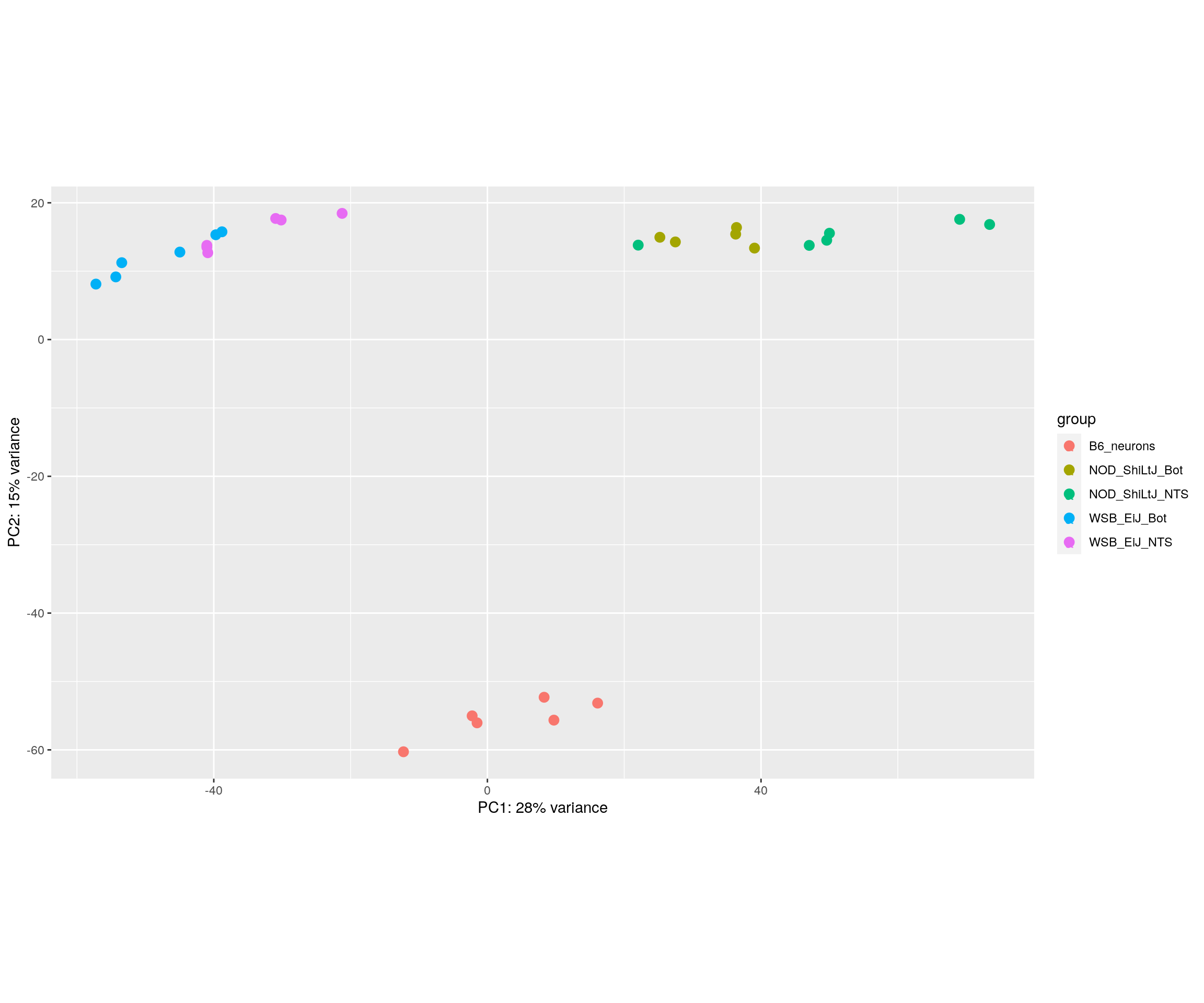
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
Differential gene expression analysis
design(ddsMat)~Strain_Tissue#Running the differential expression pipeline
res <- DESeq(ddsMat)estimating size factorsestimating dispersionsgene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesfitting model and testingresultsNames(res)[1] "Intercept"
[2] "Strain_Tissue_NOD_ShiLtJ_Bot_vs_B6_neurons"
[3] "Strain_Tissue_NOD_ShiLtJ_NTS_vs_B6_neurons"
[4] "Strain_Tissue_WSB_EiJ_Bot_vs_B6_neurons"
[5] "Strain_Tissue_WSB_EiJ_NTS_vs_B6_neurons" #comparison for Strain_Tissue_NOD_ShiLtJ_Bot_vs_B6_neurons------
fdr = 0.01
#Building the results table
res.tab <- results(res, name = "Strain_Tissue_NOD_ShiLtJ_Bot_vs_B6_neurons", alpha = 0.05)
res.tablog2 fold change (MLE): Strain Tissue NOD ShiLtJ Bot vs B6 neurons
Wald test p-value: Strain Tissue NOD ShiLtJ Bot vs B6 neurons
DataFrame with 31329 rows and 6 columns
baseMean log2FoldChange lfcSE stat
<numeric> <numeric> <numeric> <numeric>
ENSMUSG00000000001_Gnai3 1006.18087 0.124216 0.212866 0.583539
ENSMUSG00000000028_Cdc45 39.89799 -0.691388 0.955455 -0.723622
ENSMUSG00000000031_H19 27.36166 -1.198504 0.812701 -1.474718
ENSMUSG00000000037_Scml2 40.35147 -0.336759 0.570754 -0.590024
ENSMUSG00000000049_Apoh 5.54456 -0.131943 0.976122 -0.135171
... ... ... ... ...
ENSMUSG00000118643_AC163703.1 2.03021 -2.906548 2.328322 -1.24834
ENSMUSG00000118646_AC160405.1 3.36031 2.214392 1.262824 1.75352
ENSMUSG00000118651_CT030740.1 11.51722 0.103945 0.713464 0.14569
ENSMUSG00000118653_AC159819.1 2.69970 -6.212679 2.698101 -2.30261
ENSMUSG00000118655_AC156032.1 6.63847 -8.206398 2.965457 -2.76733
pvalue padj
<numeric> <numeric>
ENSMUSG00000000001_Gnai3 0.559530 0.840869
ENSMUSG00000000028_Cdc45 0.469298 0.793105
ENSMUSG00000000031_H19 0.140288 0.465192
ENSMUSG00000000037_Scml2 0.555174 0.838404
ENSMUSG00000000049_Apoh 0.892477 0.972622
... ... ...
ENSMUSG00000118643_AC163703.1 0.21190506 0.5685185
ENSMUSG00000118646_AC160405.1 0.07951218 0.3466496
ENSMUSG00000118651_CT030740.1 0.88416575 0.9701334
ENSMUSG00000118653_AC159819.1 0.02130073 0.1506475
ENSMUSG00000118655_AC156032.1 0.00565175 0.0570106summary(res.tab)
out of 31329 with nonzero total read count
adjusted p-value < 0.05
LFC > 0 (up) : 1462, 4.7%
LFC < 0 (down) : 1237, 3.9%
outliers [1] : 582, 1.9%
low counts [2] : 2422, 7.7%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultstable(res.tab$padj < fdr)
FALSE TRUE
26524 1801 #We subset the results table to these genes and then sort it by the log2 fold change estimate to get the significant genes with the strongest down-regulation:
resSig <- subset(res.tab, padj < fdr)
head(resSig[order(resSig$log2FoldChange), ])log2 fold change (MLE): Strain Tissue NOD ShiLtJ Bot vs B6 neurons
Wald test p-value: Strain Tissue NOD ShiLtJ Bot vs B6 neurons
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat
<numeric> <numeric> <numeric> <numeric>
ENSMUSG00000094103_Fam177a2 197.04636 -27.2210 4.79216 -5.68033
ENSMUSG00000083929_Gm10600 23.00282 -24.6894 4.79389 -5.15018
ENSMUSG00000094065_Ccl21d 18.03219 -23.5157 4.79443 -4.90481
ENSMUSG00000105018_Gm43546 12.97464 -22.3935 4.80408 -4.66136
ENSMUSG00000108774_Gm45136 12.97464 -22.3935 4.80408 -4.66136
ENSMUSG00000112101_Gm47924 6.89118 -20.4895 4.81798 -4.25271
pvalue padj
<numeric> <numeric>
ENSMUSG00000094103_Fam177a2 1.34438e-08 7.03874e-07
ENSMUSG00000083929_Gm10600 2.60230e-07 1.02518e-05
ENSMUSG00000094065_Ccl21d 9.35188e-07 3.25420e-05
ENSMUSG00000105018_Gm43546 3.14131e-06 9.64004e-05
ENSMUSG00000108774_Gm45136 3.14131e-06 9.64004e-05
ENSMUSG00000112101_Gm47924 2.11203e-05 5.34614e-04# with the strongest up-regulation:
head(resSig[order(resSig$log2FoldChange, decreasing = TRUE), ])log2 fold change (MLE): Strain Tissue NOD ShiLtJ Bot vs B6 neurons
Wald test p-value: Strain Tissue NOD ShiLtJ Bot vs B6 neurons
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat
<numeric> <numeric> <numeric> <numeric>
ENSMUSG00000068397_Gm10240 196.5879 21.2809 1.96602 10.82435
ENSMUSG00000084383_Gm13370 196.5879 21.2809 1.96602 10.82435
ENSMUSG00000084010_Gm13302 152.1574 21.0411 2.36194 8.90840
ENSMUSG00000112012_Gm47025 100.9697 20.1941 1.84435 10.94915
ENSMUSG00000094248_Hist1h2ao 28.9568 18.5113 4.19201 4.41586
ENSMUSG00000078087_Rps12l1 19.4819 18.4044 4.77807 3.85185
pvalue padj
<numeric> <numeric>
ENSMUSG00000068397_Gm10240 2.63937e-27 1.38445e-24
ENSMUSG00000084383_Gm13370 2.63937e-27 1.38445e-24
ENSMUSG00000084010_Gm13302 5.17739e-19 1.32117e-16
ENSMUSG00000112012_Gm47025 6.70717e-28 3.72511e-25
ENSMUSG00000094248_Hist1h2ao 1.00608e-05 2.73748e-04
ENSMUSG00000078087_Rps12l1 1.17227e-04 2.40963e-03Visualization
#Volcano plot
## Obtain logical vector regarding whether padj values are less than 0.05
threshold_OE <- (res.tab$padj < fdr & abs(res.tab$log2FoldChange) >= 1)
## Determine the number of TRUE values
length(which(threshold_OE))[1] 1747## Add logical vector as a column (threshold) to the res.tab
res.tab$threshold <- threshold_OE
## Sort by ordered padj
res.tab_ordered <- res.tab %>%
data.frame() %>%
rownames_to_column(var="ENSEMBL") %>%
tidyr::separate(., ENSEMBL, c("ENSEMBL", "SYMBOL"), sep = "_") %>%
arrange(padj) %>%
mutate(genelabels = "") %>%
as_tibble()
## Create a column to indicate which genes to label
res.tab_ordered$genelabels[1:10] <- res.tab_ordered$SYMBOL[1:10]
#display res.tab_ordered
DT::datatable(res.tab_ordered[res.tab_ordered$padj < fdr,],
filter = list(position = 'top', clear = FALSE),
extensions = 'Buttons',
options = list(dom = 'Blfrtip',
buttons = c('csv', 'excel'),
lengthMenu = list(c(10,25,50,-1),
c(10,25,50,"All")),
pageLength = 40,
scrollY = "300px",
scrollX = "40px"),
caption = htmltools::tags$caption(style = 'caption-side: top; text-align: left; color:black; font-size:200% ;','DEG for NOD_ShiLtJ_Bot_vs_B6_neurons (fdr < 0.01)'))#Volcano plot
volcano.plot <- ggplot(res.tab_ordered) +
geom_point(aes(x = log2FoldChange, y = -log10(padj), colour = threshold)) +
scale_color_manual(values=c("blue", "red")) +
geom_text_repel(aes(x = log2FoldChange, y = -log10(padj),
label = genelabels,
size = 3.5)) +
ggtitle("DEG for NOD_ShiLtJ_Bot_vs_B6_neurons") +
xlab("log2 fold change") +
ylab("-log10 adjusted p-value") +
theme(legend.position = "none",
plot.title = element_text(size = rel(1.5), hjust = 0.5),
axis.title = element_text(size = rel(1.25)))
print(volcano.plot)Warning: Removed 3004 rows containing missing values (geom_point).Warning: Removed 3004 rows containing missing values (geom_text_repel).Warning: ggrepel: 6 unlabeled data points (too many overlaps). Consider
increasing max.overlaps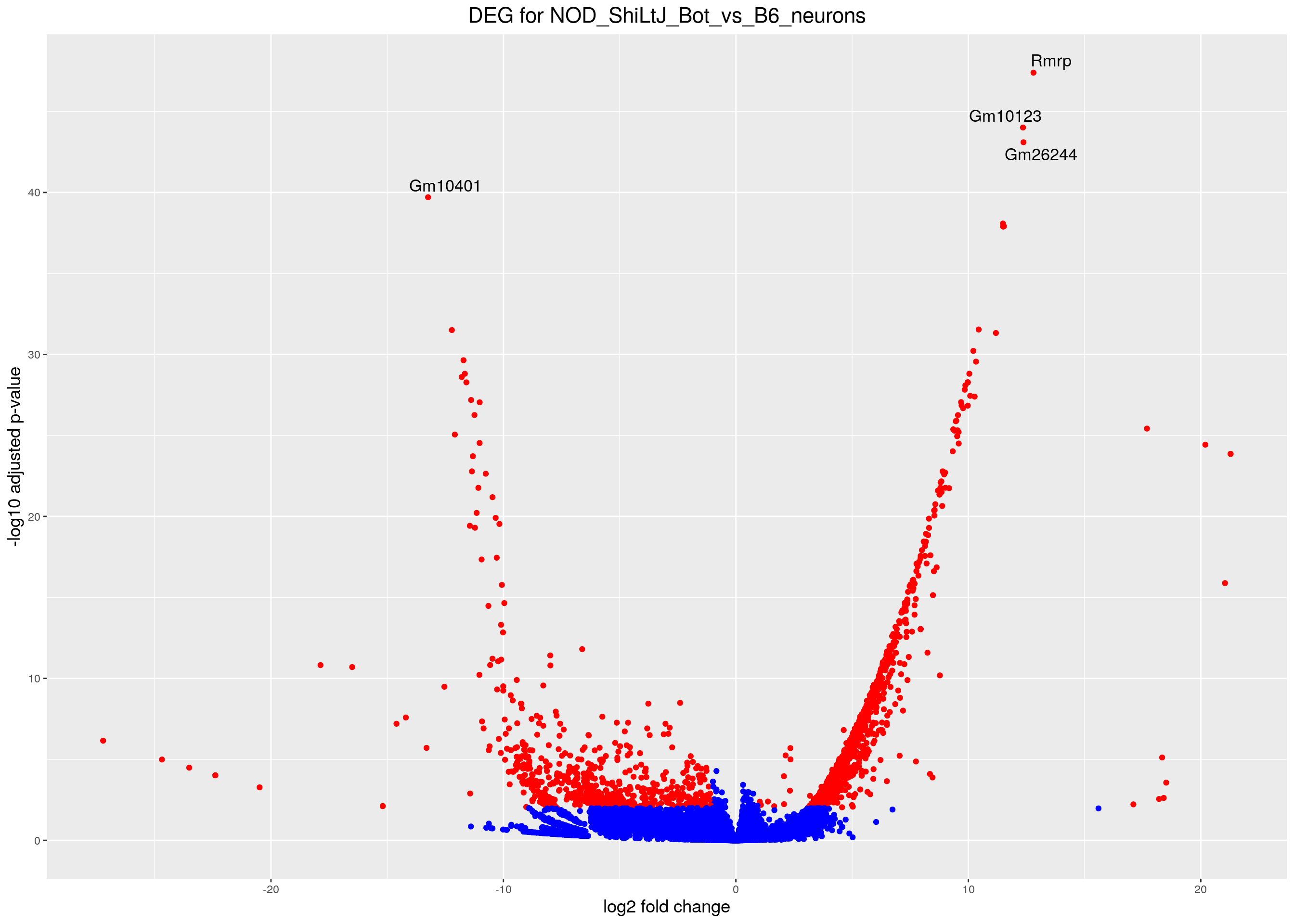
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
#heatmap
# Extract normalized expression for significant genes fdr < 0.000001 & abs(log2FoldChange) >= 1)
normalized_counts_sig <- normalized_counts %>%
filter(gene %in% rownames(subset(resSig, padj < 0.000001 & abs(log2FoldChange) >= 1)))
#Set a color palette
heat_colors <- brewer.pal(6, "YlOrRd")
#Run pheatmap using the metadata data frame for the annotation
pheatmap(as.matrix(normalized_counts_sig[,-1]),
color = heat_colors,
cluster_rows = T,
show_rownames = F,
annotation_col = df,
border_color = NA,
fontsize = 10,
scale = "row",
fontsize_row = 10,
height = 20,
main = "Heatmap of the top DEGs in NOD_ShiLtJ_Bot_vs_B6_neurons (fdr < 0.000001 & abs(log2FoldChange) >= 1)")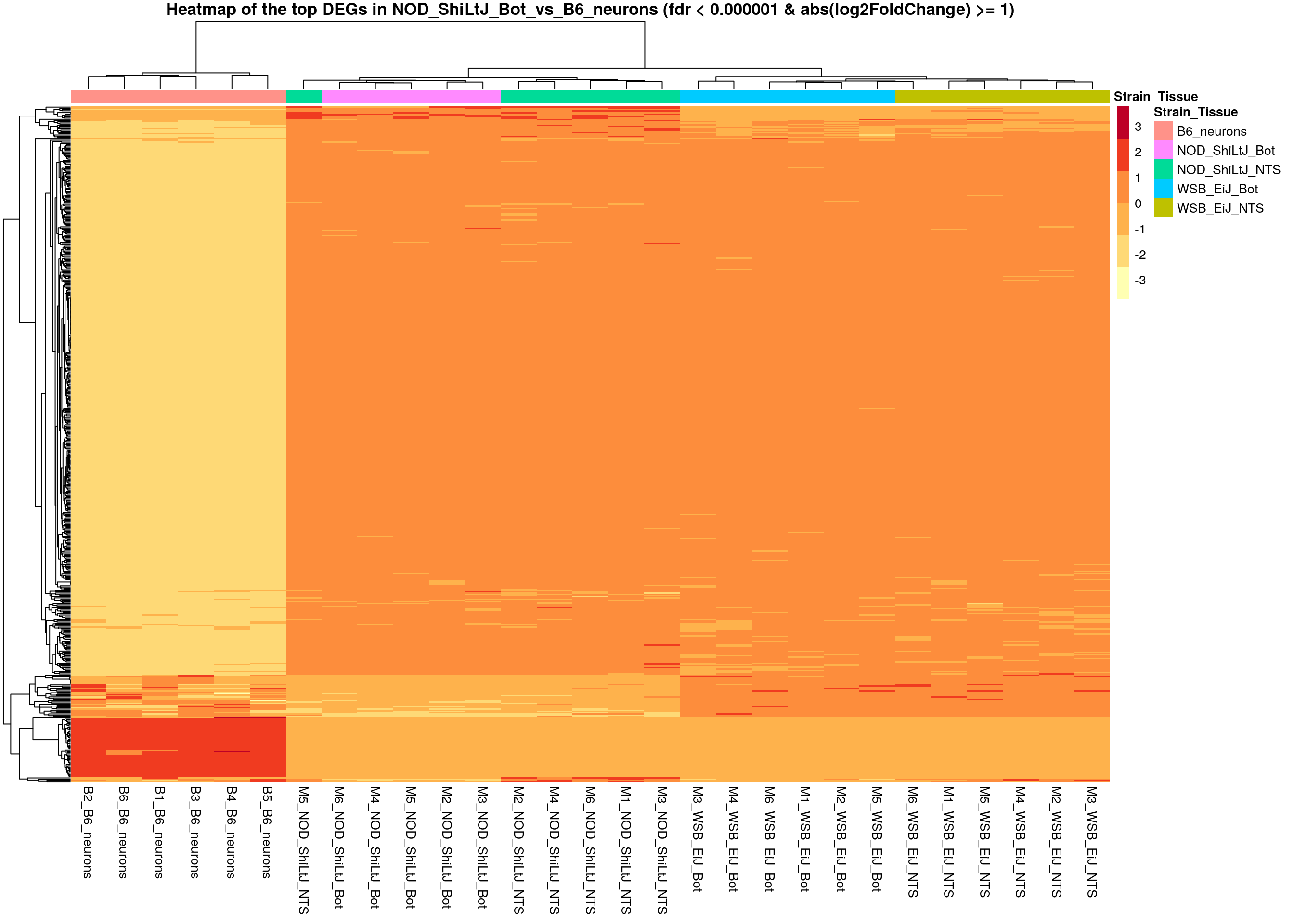
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
Enrichment
#enrichment analysis
dbs <- c("WikiPathways_2019_Mouse",
"GO_Biological_Process_2021",
"GO_Cellular_Component_2021",
"GO_Molecular_Function_2021",
"KEGG_2019_Mouse",
"Mouse_Gene_Atlas",
"MGI_Mammalian_Phenotype_Level_4_2019")
#results------
resSig.tab <- resSig %>%
data.frame() %>%
rownames_to_column(var="ENSEMBL") %>%
as_tibble() %>%
tidyr::separate(., ENSEMBL, c("ENSEMBL", "SYMBOL"), sep = "_")
#up-regulated tissue genes---------------------------
up.genes <- resSig.tab %>%
filter(log2FoldChange > 0) %>%
pull(SYMBOL)
#up-regulated genes enrichment
up.genes.enriched <- enrichr(as.character(na.omit(up.genes)), dbs)Uploading data to Enrichr... Done.
Querying WikiPathways_2019_Mouse... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Querying KEGG_2019_Mouse... Done.
Querying Mouse_Gene_Atlas... Done.
Querying MGI_Mammalian_Phenotype_Level_4_2019... Done.
Parsing results... Done.for (j in 1:length(up.genes.enriched)){
up.genes.enriched[[j]] <- cbind(data.frame(Library = names(up.genes.enriched)[j]),up.genes.enriched[[j]])
}
up.genes.enriched <- do.call(rbind.data.frame, up.genes.enriched) %>%
filter(Adjusted.P.value <= 0.1) %>%
mutate(logpvalue = -log10(P.value)) %>%
arrange(desc(logpvalue))
#display up.genes.enriched
DT::datatable(up.genes.enriched,filter = list(position = 'top', clear = FALSE),
options = list(pageLength = 40, scrollY = "300px", scrollX = "40px"))#barpot
up.genes.enriched.plot <- up.genes.enriched %>%
filter(Adjusted.P.value <= 0.1) %>%
mutate(Term = fct_reorder(Term, -logpvalue)) %>%
ggplot(data = ., aes(x = Term, y = logpvalue, fill = Library, label = Overlap)) +
geom_bar(stat = "identity") +
geom_text(position = position_dodge(width = 0.9),
hjust = 0) +
theme_bw() +
ylab("-log(p.value)") +
xlab("") +
ggtitle("Enrichment of up-regulated tissue genes \n(Adjusted.P.value <= 0.1)") +
theme(plot.background = element_blank() ,
panel.border = element_blank(),
panel.background = element_blank(),
#legend.position = "none",
plot.title = element_text(hjust = 0),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank()) +
theme(axis.line = element_line(color = 'black')) +
theme(axis.title.x = element_text(size = 12, vjust=-0.5)) +
theme(axis.title.y = element_text(size = 12, vjust= 1.0)) +
theme(axis.text = element_text(size = 12)) +
theme(plot.title = element_text(size = 12)) +
coord_flip()
up.genes.enriched.plot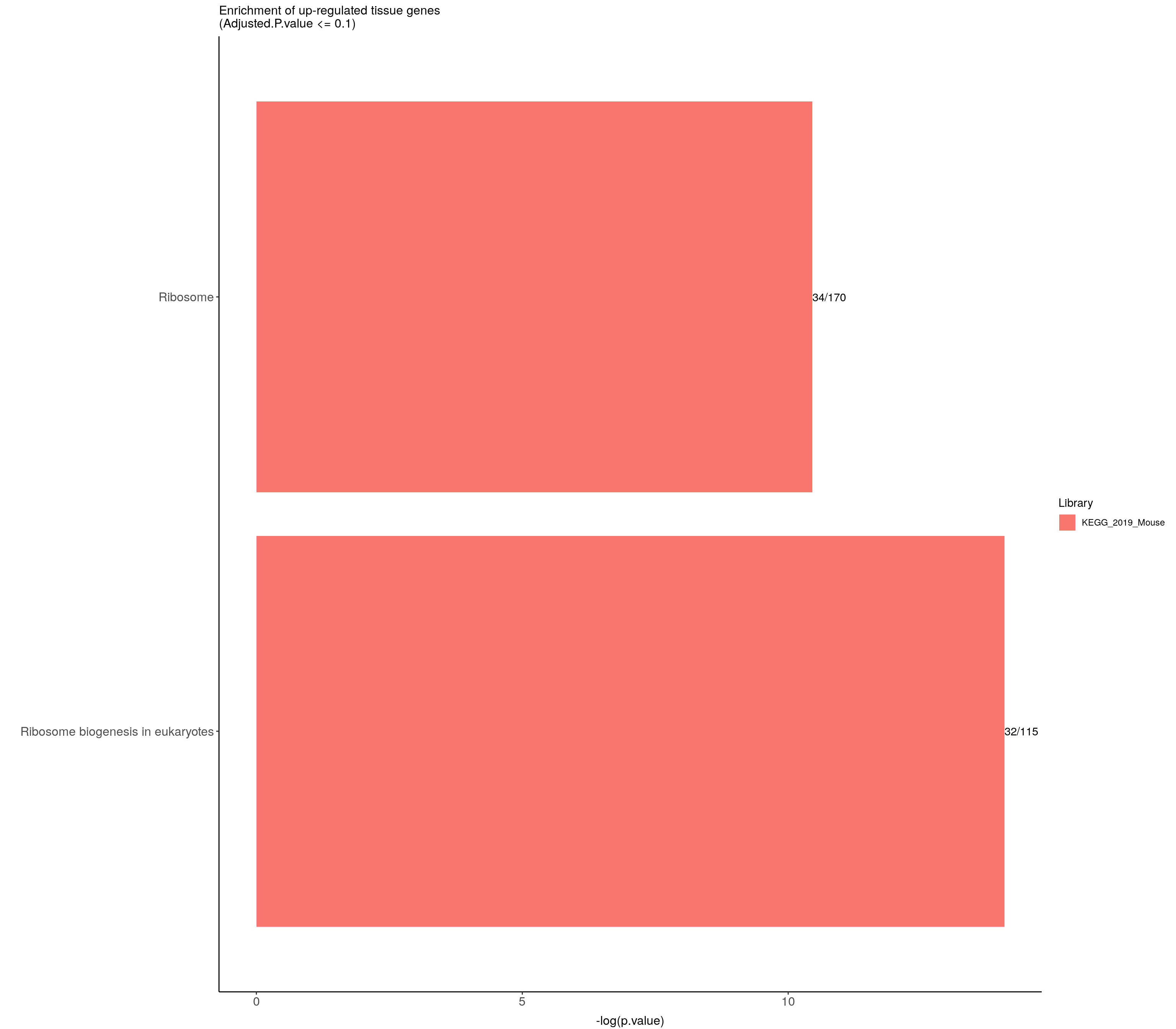
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
#down-regulated tissue genes---------------------------
down.genes <- resSig.tab %>%
filter(log2FoldChange < 0) %>%
pull(SYMBOL)
#down-regulated genes enrichment
down.genes.enriched <- enrichr(as.character(na.omit(down.genes)), dbs)Uploading data to Enrichr... Done.
Querying WikiPathways_2019_Mouse... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Querying KEGG_2019_Mouse... Done.
Querying Mouse_Gene_Atlas... Done.
Querying MGI_Mammalian_Phenotype_Level_4_2019... Done.
Parsing results... Done.for (j in 1:length(down.genes.enriched)){
down.genes.enriched[[j]] <- cbind(data.frame(Library = names(down.genes.enriched)[j]),down.genes.enriched[[j]])
}
down.genes.enriched <- do.call(rbind.data.frame, down.genes.enriched) %>%
filter(Adjusted.P.value <= 0.2) %>%
mutate(logpvalue = -log10(P.value)) %>%
arrange(desc(logpvalue))
#display down.genes.enriched
DT::datatable(down.genes.enriched,filter = list(position = 'top', clear = FALSE),
options = list(pageLength = 40, scrollY = "300px", scrollX = "40px"))#barpot
down.genes.enriched.plot <- down.genes.enriched %>%
filter(Adjusted.P.value <= 0.2) %>%
mutate(Term = fct_reorder(Term, -logpvalue)) %>%
ggplot(data = ., aes(x = Term, y = logpvalue, fill = Library, label = Overlap)) +
geom_bar(stat = "identity") +
geom_text(position = position_dodge(width = 0.9),
hjust = 0) +
theme_bw() +
ylab("-log(p.value)") +
xlab("") +
ggtitle("Enrichment of down-regulated tissue genes \n(Adjusted.P.value <= 0.2)") +
theme(plot.background = element_blank() ,
panel.border = element_blank(),
panel.background = element_blank(),
#legend.position = "none",
plot.title = element_text(hjust = 0),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank()) +
theme(axis.line = element_line(color = 'black')) +
theme(axis.title.x = element_text(size = 12, vjust=-0.5)) +
theme(axis.title.y = element_text(size = 12, vjust= 1.0)) +
theme(axis.text = element_text(size = 12)) +
theme(plot.title = element_text(size = 12)) +
coord_flip()
down.genes.enriched.plot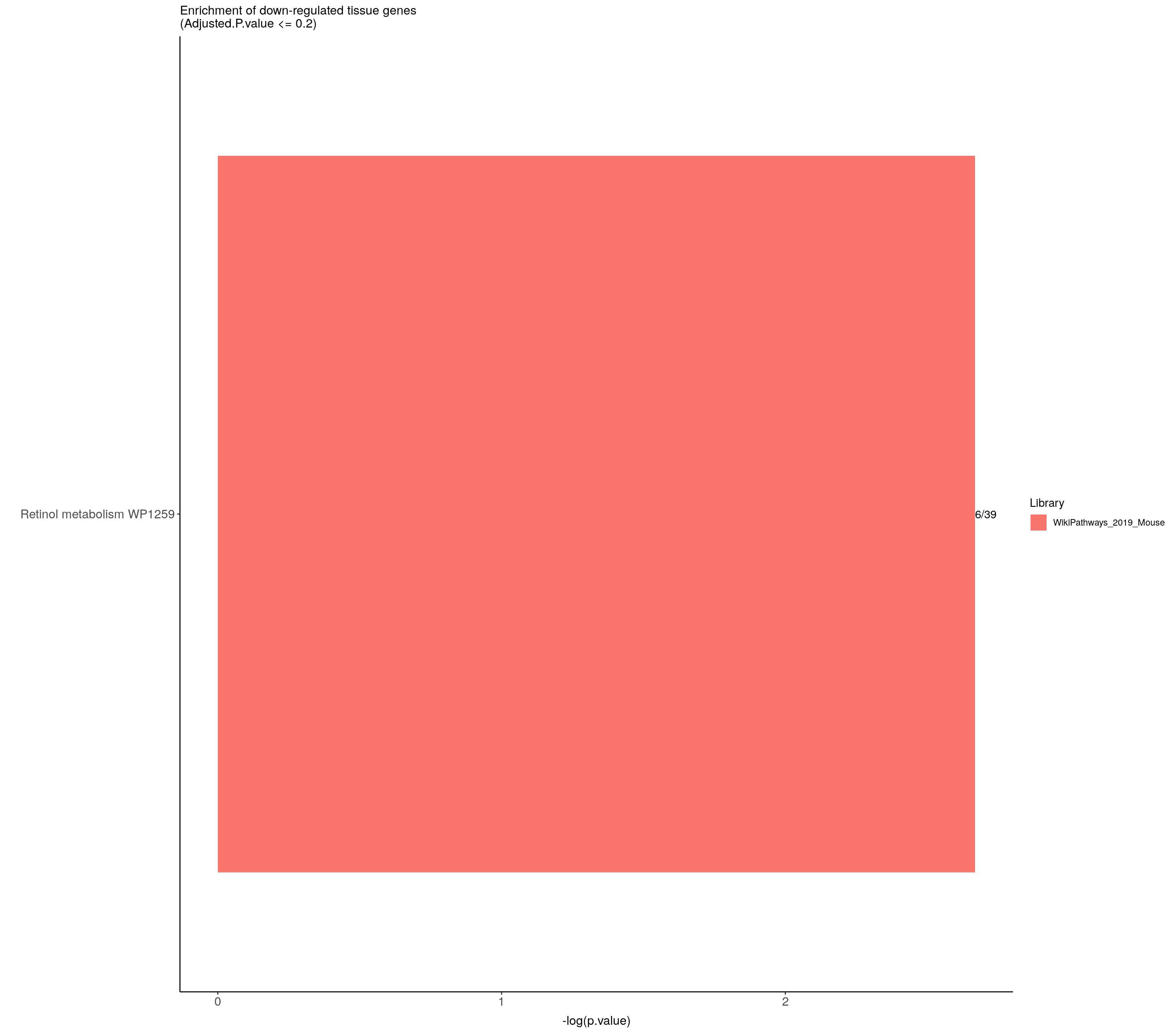
| Version | Author | Date |
|---|---|---|
| bc47d57 | xhyuo | 2022-09-29 |
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.2 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] sva_3.38.0 BiocParallel_1.24.1
[3] genefilter_1.72.1 mgcv_1.8-34
[5] nlme_3.1-152 ggplotify_0.0.5
[7] cowplot_1.1.1 enrichR_3.0
[9] DT_0.17 ggrepel_0.9.1
[11] pheatmap_1.0.12 DESeq2_1.30.1
[13] SummarizedExperiment_1.20.0 MatrixGenerics_1.2.1
[15] matrixStats_0.58.0 GenomicRanges_1.42.0
[17] GenomeInfoDb_1.26.7 RColorBrewer_1.1-2
[19] org.Mm.eg.db_3.12.0 AnnotationDbi_1.52.0
[21] IRanges_2.24.1 S4Vectors_0.28.1
[23] Biobase_2.50.0 BiocGenerics_0.36.1
[25] gplots_3.1.1 Glimma_2.0.0
[27] edgeR_3.32.1 limma_3.46.0
[29] forcats_0.5.1 dplyr_1.0.4
[31] purrr_0.3.4 readr_1.4.0
[33] tidyr_1.1.2 tibble_3.0.6
[35] ggplot2_3.3.3 tidyverse_1.3.0
[37] stringr_1.4.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] colorspace_2.0-0 rjson_0.2.20 ellipsis_0.3.1
[4] rprojroot_2.0.2 XVector_0.30.0 fs_1.5.0
[7] rstudioapi_0.13 farver_2.0.3 bit64_4.0.5
[10] lubridate_1.7.9.2 xml2_1.3.2 splines_4.0.3
[13] cachem_1.0.4 geneplotter_1.68.0 knitr_1.31
[16] jsonlite_1.7.2 broom_0.7.4 annotate_1.68.0
[19] dbplyr_2.1.0 BiocManager_1.30.10 compiler_4.0.3
[22] httr_1.4.2 rvcheck_0.1.8 backports_1.2.1
[25] assertthat_0.2.1 Matrix_1.3-2 fastmap_1.1.0
[28] cli_2.3.0 later_1.1.0.1 htmltools_0.5.1.1
[31] tools_4.0.3 gtable_0.3.0 glue_1.4.2
[34] GenomeInfoDbData_1.2.4 Rcpp_1.0.6 cellranger_1.1.0
[37] vctrs_0.3.6 crosstalk_1.1.1 xfun_0.21
[40] rvest_0.3.6 lifecycle_1.0.0 gtools_3.8.2
[43] XML_3.99-0.5 zlibbioc_1.36.0 scales_1.1.1
[46] hms_1.0.0 promises_1.2.0.1 curl_4.3
[49] yaml_2.2.1 memoise_2.0.0 stringi_1.5.3
[52] RSQLite_2.2.3 highr_0.8 caTools_1.18.1
[55] rlang_1.0.2 pkgconfig_2.0.3 bitops_1.0-6
[58] evaluate_0.14 lattice_0.20-41 labeling_0.4.2
[61] htmlwidgets_1.5.3 bit_4.0.4 tidyselect_1.1.0
[64] magrittr_2.0.1 R6_2.5.0 generics_0.1.0
[67] DelayedArray_0.16.3 DBI_1.1.1 pillar_1.4.7
[70] haven_2.3.1 whisker_0.4 withr_2.4.1
[73] survival_3.2-7 RCurl_1.98-1.2 modelr_0.1.8
[76] crayon_1.4.1 KernSmooth_2.23-18 rmarkdown_2.6
[79] locfit_1.5-9.4 grid_4.0.3 readxl_1.3.1
[82] blob_1.2.1 git2r_0.28.0 reprex_1.0.0
[85] digest_0.6.27 xtable_1.8-4 httpuv_1.5.5
[88] gridGraphics_0.5-1 munsell_0.5.0 This R Markdown site was created with workflowr